I. Introduction
1) Cardiomyocytes
Le muscle cardiaque est constitué de cellules, les cardiomyocytes, liés les uns aux autres par des disques intercalaires. Les embranchements des cardiomyocytes forment un réseau complexe tridimentionnel. Tous les cardiomyocytes sont capables de dépolarisations rythmiques spontanées. Mais, c'est un groupe de myocytes de l'atrium qui constitue le pacemaker et propage les stimulations électriques à travers le myocarde par des gap junctions situées entre les myocytes. C'est pourquoi, malgré le fait qu'il soit constitué par des cellules séparées, le myocarde se comporte comme un syncytium [1]. Le cardiomyocyte, en plus de sa fonction contractile propre, possède des propriétés endocrines en ce sens qu'il est capable de synthétiser et libérer dans son environnement immédiat et dans la circulation générale des facteurs autacoïdes tels que certaines prostaglandines, comme la prostaglandine E2 et la prostacycline, des dérivés de phospholipides membranaires et des hormones peptidiques, les peptide natriurétique auriculaire ou ANP et le BNP (Brain natriuretic peptide).
Le cardiomyocyte possède dans sa membrane cellulaire des récepteurs à plusieurs hormones : les catécholamines, l'endothéline, l'angiotensine ll et la vasopressine, l'aldostérone, des hormones thyroïdiennes, l'insuline entre autres. L'activation des récepteurs à l'angiotensine ll (Ang II) et à la vasopressine aboutit à des effets variés, comprenant des effets inotropes et chronotropes, des effets de croissance par l'intermédiaire de la stimulation d'oncogènes ainsi qu'à la stimulation de la fonction endocrine des cardiomyocytes. L'angiotensine ll et l'endothéline-1 peuvent provoquer une réponse hypertrophique chez le rat [2], une réponse qu'on observe également in vivo durant le développement du coeur. En effet, dans la période post-natale, les cellules musculaires cardiaques perdent leur capacité à se diviser et dès ce moment, la croissance du coeur se fait par hypertrophie : phénomène caractérisé par une augmentation de la quantité de protéine par cellule [3].
2) Prostaglandines
2.1) Sécrétion de prostacyclines dans des cultures de cardiomyocytes
L'augmentation de la sécrétion de prostacycline (PGI2) dans le coeur après des lésions ischémiques peut résulter de la production de PGI2 par différents types de cellules, notamment les cellules endothéliales. Des expériences effectuées dans des cellules soumises d'abord à l'ischémie puis à une réoxygénation ont montré une libération de prostacycline plus grande lors de la réoxygénation [4], [5]. Des travaux effectués par notre laboratoire ont montré une augmentation de la sécrétion de prostacycline par l'Ang ll et la vasopressine (AVP) dans les cardiomyocytes ventriculaires, par la voie de l'activation de la PKC et la phospholipase A2 [6, 7]
La synthèse de prostacycline est régulée par deux étapes limitantes : la libération d'acide arachidonique des phospholipides membranaires par la phospholipase A2 (PLA2) et la conversion de cet acide arachidonique en précurseurs de prostanoïdes (PGH2) par la cyclooxygénase.
2.2) Phospholipase A2
La phospholipase A2 est impliquée dans différentes réponses cellulaires, dont la défense de l'hôte, la coagulation, la digestion. De nombreuses études effectuées dans différents types cellulaires ont montré que la PLA2 joue un rôle clé dans la régulation des voies de transduction des signaux médié par les lipides.
Il existe trois types de cet enzyme :
- La PLA2 sécrétoire qui demande du Ca++ en concentration millimolaire pour accomplir sa fonction enzymatique. Elle est elle-même subdivisée en deux sous-groupes basés sur sa sélectivité tissulaire. On trouve le groupe 1 principalement dans le pancréas. Et le groupe dans les tissus non pancréatiques [8].
- La PLA2 cytosolique qui requiert du Ca++ en concentration submicromolaire pour une activité cytosolique optimale. On la trouve dans le compartiment cytosolique de la plupart des cellules [9].
- La PLA2 qui est indépendante du Ca++.
2.3) Cyclooxygénase
Le Prostaglandine endoperoxyde H synthase ou cyclooxygénase catalyse une étape de la formation des prostaglandines et du thromboxane. Elle possède deux fonctions enzymatiques : une fonction dite cyclooxygénase, reponsable de l'oxydation de l'acide arachidonique en PGG2 suite à l'insertion de deux molécules d'O2 et une fonction peroxydase qui réduit les PGG2 en PGH2 [10]. Il existe 2 types de PGHS : PGHS-1 et PGHS-2 ou COX-1 et COX-2 qui catalysent toutes les deux la formation des PGH2 à partir de l'acide arachidonique.
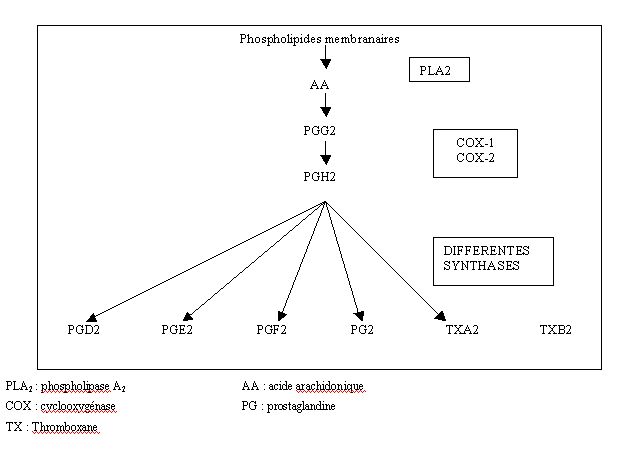
La PLA2 et la COX transforment les phospholipides membranaires en PGH2, qui est ensuite converti par des enzymes spécifiques des tissus en prostacycline, prostaglandines et thromboxane. La PGH2, après avoir été synthétisé par les COX, est transformé par des synthases différentes en PGD2, PGE2, PGF2a ou Txa2
Les deux isoenzymes de la cyclooxygénase possèdent essentiellement la même structure primaire avec 60% d'identité ainsi qu'une structure cristallographique semblable [11, 12] et elles possèdent des propriétés cinétiques très proches. Les deux sont inhibées par des antiinflammatoires non stéroïdiens (AINS) bien que des inhibiteurs spécifiques de chacune aient été développés. COX-1 et -2 se trouvent dans les cellules sous forme d'homodimères attachés par leur domaines hydrophobes aux membranes du réticulum endoplasmique et du noyau [13, 14]. Malgré leurs points communs structuraux et fonctionnels, les deux isoformes possèdent des fonctions biologiques différentes.
COX-1 et COX-2 sont codées par des gènes distincts [15].
La COX-1 est considérée comme l'enzyme constitutive ; et son expression est régulée tout au long de son développement ; elle est responsable de la production physiologique, constitutive de prostaglandines. Une fois que l'enzyme a formé les prostanoïdes dans le réticulum endoplasmique, ces derniers agissent par des récepteurs de surface, pour effectuer des tâches d'entretien de l'organisme ; par exemple, l'équilibre hydrosodé par le rein, ou la protection gastrique contre l'acidité, ou encore l'hémostase.
La COX-2 au contraire est l'enzyme dite inductible, qui dans des condition standards est habituellement absente des cellules et qui est exprimée de manière transitoire en réponse à des facteurs de croissance, des cytokines (Il-1, IL-6, PDGF, TNF-a) ou des promoteurs de tumeur ; elle prédomine dans les conditions inflammatoires. La COX-2 synthétise les prostanoïdes sur l'enveloppe nucléaire et ces derniers semblent agir au niveau nucléaire durant la réplication ou la différenciation nucléaire [16].
Les éicosanoïdes ont des effets biologiques variés malgré leur structures semblables. Ils sont impliqués dans les fonctions respiratoires, puisqu'ils provoquent une bronchoconstriction. Les PGE2 stimulent la production de mucus gastrique qui avec les bicarbonates inhibent la rétrodiffusion des H+. En outre ils tempèrent la sécrétion acide de l'estomac par une action directe sur les cellules pariétales. Les PGE2 et les PGI2 permettent une vasodilatation rénale. Les PGI2 permettent une relaxation des cellules musculaires lisses vasculaires (VSMC), mais provoquent une contraction des cellules musculaires lisses du tractus digestif. Ils sont en outre connus pour leur effet proinflammatoire. On utilise depuis le début du siècle les antiinflammatoires non stéroïdiens, qui inhibent les deux isoenzymes et empêchent ainsi la production de prostaglandines et de thromboxanes proinflammatoires [16]. Mais cette supression complète d'eicosanoïdes est à l'origine de nombreux effets indésirables de ces médicaments, notamment l'insuffisance rénale et l'ulcère gastrique. C'est pourquoi on développe de plus en plus des inhibiteurs spécifiques de la COX-2. Ainsi, l'inflammation est diminuée, mais la production basale d'eicosanoïdes nécessaire au maintien de l'homéostasie gastrique et rénale est conservée [17, 18].
Les COX ont en outre un intérêt thérapeuthique certain au vu de leur implication dans les thromboses coronaires , dans l'inflammation, dans le cancer du côlon et dans la maladie d'Alzheimer [19].
Les premières études considéraient la COX-2 comme délétère tandis que la COX-1 était vue comme protectrice, mais actuellement, la distinction n'est plus aussi claire.
Plusieurs études récentes ont montré des effets variés de la COX-2 au niveau du coeur.
Adderley et al. (1999) [20] ont montré que les lésions des cardiomyocytes dues au H2O2 et à la doxorubicine sont limitées par la formation de prostacycline, qui reflète l'induction de COX-2.
De même, Noreen P. Dowd ont montré que le degré de lésion cardiaque chez un animal traité avec de la doxorubicine et du SC236 était diminué par l'administration préalable d'iloprost, un analogue de la PGI2 [21].
Une étude canadienne [22] a démontré l'induction de la COX-2, en association au processus inflammatoire et à la cicatrisation, dans le myocarde lors de l'infarctus du myocarde . En revanche, le rôle exact de cet enzyme dans cette maladie fatale n'a pas été déterminé [22].
Une étude menée chez le lapin a montré que l'augmentation de la COX-2 joue un rôle essentiel dans la protection du coeur lors de la phase tardive du préconditionnement ischémique (cycle d'occlusion/reperfusion coronaire de 4 minutes). En effet, dans cette expérience, le préconditionnement ischémique montre une augmentation rapide de l'ARMm de la COX-2, une induction importante de la COX-2 dans les 24 heures et une augmentation du contenu cardiaque en prostaglandines, produits enzymatiques de la COX-2. Dans cette expérience, l'utilisation d'inhibiteurs sélectifs de la COX-2 (NS-398 et celecoxib) bloquent totalement l'effet cardioprotecteur, tant pour les lésions ischémiques réversibles (ischémie), que pour les lésions irréversibles (infarctus) Cette étude semble donc définir la COX-2 comme un enzyme cardioprotecteur [23].
Une augmentation de l'enzyme COX-2 ainsi que de son mRNA a été démontré lors d'un rejet d'allogreffe cardiaque. Néanmoins, l'inflammation et l'apoptose des cardiomyocytes n'ont pas été réduits de manière significative par un traitement d'inhibiteurs de la COX-2 [24].
Une fibrose cardiaque a été rapporté par Wong et al (1988) [25] chez la souris dont le gène de la COX-2 a été inactivé, suggérant que la COX-2 aurait des propriétés cardioprotectrices.
3) Angiotensine II (Ang ll)
L'Ang ll est un octapeptide et le composant actif principal du système rénine-angiotensine. Ce système est important pour le contrôle de la pression artérielle et l'équilibre ionique, par l'effet direct de l'Ang ll et par l'intermédiaire de la sécrétion d'aldostérone. La diminution de la pression artérielle moyenne de même qu'une augmentation du K+ plasmatique ou une restriction sodée stimule respectivement les barorécepteurs ou les chémorécepteurs de l'appareil juxtaglomérulaire du rein, ce qui provoque la sécrétion de la rénine, un enzyme de la zone de la Macula Densa, qui agit pendant 30 minutes à une heure en transformant l'angiotensinogène en angiotensine l au niveau du foie. Ensuite, l'enzyme de conversion de l'angiotensine convertit l'angiotensine l en angiotensine ll au niveau des poumons ; cet enzyme est détruit en quelques minutes dans le sang par l'action d'angiotensinogénases. L'Ang ll permet une constriction artérielle directe, la réabsorption rénale de Na+, une augmentation de la précharge cardiaque, de l'inotropisme et du chronotropisme ainsi qu'une stimulation de la soif. L'effet final est une augmentation du volume plasmatique et de la résistance vasculaire périphérique, donc une augmentation de la pression artérielle. L'Ang ll permet également une augmentation de la sécrétion d'aldostérone par la zone glomérulaire du cortex surrénalien. L'aldostérone, un minéralocorticoïde, qui au niveau rénal augmente la réabsorption du sodium, entraîne donc une rétention d'eau, mécanisme qui permet également une augmentation de la pression artérielle par augmentation de la précharge cardiaque, mais de manière moins importante que l'angiotensine elle-même. En outre, l'aldostérone provoque une augmentation de l'excrétion du K+. Ensuite, un rétrocontrôle négatif permet un arrêt de la sécrétion de rénine, ce qui permet une stabilisation de la pression artérielle et de l'équilibre ionique [26].
Le rôle de l'Ang ll a été étudié dans plusieurs pathologies cardiaques en utilisant des inhibiteurs du système rénine-angiotensine (RA). Dans nombre de ces études cliniques, l'efficacité des inhibiteurs de l'enzyme de conversion de l'angiotensine dans l'hypertension artérielle et l'insuffisance cardiaque a été démontrée [27, 28]. Habituellement, ce système rénine-angiotensine est décrit comme un système général avec une Ang ll distribuée dans tout le corps. Pourtant, il existerait d'après plusieurs auteurs [29]., des systèmes RA localisés dans des organes spécifiques comme par exemple le coeur qui fonctionne par voie autocrine ou paracrine.
L'Ang ll se lie à deux types de récepteurs AT1 et AT2 qui sont exprimés dans de nombreux tissus dont le coeur [30]. Ces récepteurs appartiennent à une famille de récepteurs à 7 domaines transmemembranaires et se distinguent les uns des autres par leur affinité pour les antagonistes non peptidiques [31]. Ces récepteurs sont augmentés après un infarctus du myocarde [32] et une hypertrophie [33]. Dans le coeur, ce sont les récepteurs AT1 qui sont responsables de la plupart des effets physiologiques de l'hormone [34]. D'après une étude menée aux Pays-bas [35]., il semble que lors d'hypertrophie cardiaque due à des stress physiques, l'Ang ll, ainsi que l'endothéline-1 et le TGF-b libérés par les cellules cardiaques et vasculaires participeraient à ce phénomène par une action autocrine et paracrine.
4) Endothéline-1 (ET-1)
La famille des endothélines comporte trois membres : ET-1, ET-2, ET-3, qui ont été identifiés par clonage [36]. Une préproendothéline est clivée par des endopeptidases spécifiques en grande endothéline, puis l'enzyme de conversion de l'endothéline la convertit en endothéline.
L'ET-1 est un grand peptide qui compte 21 acides aminés. La transcription de son gène peut être régulée dans la cellule endothéliale au niveau de l'ARNm par la thrombine, l'adrénaline, l'Ang ll, l'ADH, le TGF-b, l'ester de phorbol ; sa libération peut être inhibée par le NO et l'ANP [37]. On la trouve à la surface des cellules endothéliales de la plupart des vaisseaux. Le stimulus nécessaire à sa sécrétion est la lésion de l'endothélium vasculaire. Le rôle physiologique de cette hormone n'est pas encore élucidé, mais on la trouve augmentée dans certaines pathologies, comme la toxémie gravidique, l'insuffisance rénale aiguë ou chronique [38]. L'endothéline administrée de manière exogène a montré une augmentation de la résistance vasculaire périphérique. Mais durant les premières minutes d'administration intraveineuse, l'endothéline diminue la résistance vasculaire périphérique ; probablement dû à une libération de composés vasodilatateurs comme le NO, le PGH2 ou l'ANP [37].
On a découvert que l'ET-1 possédait un rôle de facteur de croissance dans certaines cellules de mammifères, comme les VSMC et les fibroblastes [39, 40]. En ce qui concerne le coeur, Kojima et al., 1995 [41] ont démontré qu'un antagoniste non spécifique du récepteur à l'endothéline diminuait la taille de l'infarctus chez le rat, le lapin, le chien, quand on l'administre avant ou après un infarctus aigu du myocarde. L'ET-1 a également un effet inotrope positif dans des cardiomyocytes ventriculaires en culture [41], en outre, l'ET-1 induit une hypertrophie des cardiomyocytes [42, 43, 44]. L'ET-1 est également produite par des cellules non endothéliales comme les VSMC, les cellules épithéliales rénales, les cellules mésangiales glomérulaires et certaines lignées de cellules cancéreuses [45, 46, 47, 48]. Ces observations ont mené à l'hypothèse que l'endothéline endogène produite par les cardiomyocytes pourrait être impliquée dans la pathogenèse de l'hypertrophie cardiaque par un mécanisme autocrine et paracrine. Dans ce contexte, Ito et al. (1996) [49] ont rapporté une augmentation de l'ARNm de la préproendothéline-1 et de la libération de l'ET-1 et ils ont suggéré que cette activation endogène peut, au moins en partie, médier la réponse hypertrophique induite par l'hypoxie. La synthèse myocardique accélérée d'ET-1 contribue également à la dysfonction du ventricule gauche durant la transition de la phase d'hypertrophie du ventricule gauche à celle d'insuffisance cardiaque congestive chez le rat hypertendu [50]. En outre, Kobayashi et al., (1999) [51] ont rapporté que le niveau de l'ARNm de la préproendothéline, celui de l'ET-1 ainsi que celui du récepteur étaient significativement plus hauts chez les rats en insuffisance cardiaque chronique que chez les rats contrôles.
5) Aldostérone
L'aldostérone est un minéralocorticoïde très puissant qui est responsable du le 90% de l'activité de tous les minéralocorticoïdes. L'absence de sa sécrétion par le cortex surrénalien entraînerait la mort en jours à deux semaines. Sans minéralocorticoïdes, la concentration plasmatique de K+ augmenterait à l'inverse du Na+ et du volume liquidien extracellulaire. En effet, l'aldostérone agit sur l'échangeur Na+/K+ des cellules épithéliales tubulaires rénales.
La sécrétion d'aldostérone est stimulée par : 1) L'augmentation du K+ extracellulaire ; 2) L'activation du système RA ; 3) la sécrétion basale d'ACTH, indispensable, mais qui ne provoque que peu de variation dans le taux sécrété d'aldostérone. La sécrétion d'aldostérone est diminuée en présence d'une hypernatrémie.
L'aldostérone permet par son action d'augmenter la tension artérielle et de diminuer le K+, d'effectuer un rétrocontrôle négatif pour atteindre un équilibre tensionnel et ionique [52].
Comme d'autres hormones stéroïdes, l'aldostérone commence son action en se liant à ses récepteurs intracellulaires qui ensuite sont capables de contrôler la transcription de plusieurs gènes. Dans les cardiomyocytes de rats nouveau-nés et adultes, des concentrations physiologiques d'aldostérone ont montré une augmentation de trois fois des isoformes principales des pompes Na+/K+- ATPase [52b]. En outre, dans les cellules cardiaques de rats nouveau-nés, un traitement de 24 heures d'aldostérone stimule l'activité de l'antiport Na+/H+ et de l'échangeur Cl-/HCO3- [53]. Récemment, il a été rapporté dans les cardiomyocytes de rats adultes, que l'aldostérone induisait une expression augmentée du courant de Ca2+ [54]. L'événement initial de l'action de l'aldostérone est sa liaison à son récepteur. Ce récepteur a la même affinité pour l'aldostérone et pour les glucocorticoïdes. C'est pourquoi la sélectivité in vivo de ce récepteur requiert la présence d'un enzyme, le 11b-hydrostéroïde déshydrogénase, qui métabolise les glucocorticoïdes en dérivés inactifs. Lombès et al (1995) [55] ont montré une coexpression de cet enzyme et du récepteur à l'aldostérone dans le coeur, qui contient donc les outils cellulaires nécessaires à l'action de l'aldostérone.
Un excès chronique d'aldostérone est associé à un dépôt de collagène interstitiel cardiaque et périvasculaire marqués [56, 57, 58]. La spironolactone, antagoniste de l'aldostérone, prévient cette fibrose cardiaque, ce qui indique qu'un récepteur aux minéralocorticoïdes est impliqué dans cette réponse [56]. Une étude récente nous montre l'implication de l'angiotensine-2 et de son récepteur dans le mécanisme de la fibrose cardiaque, suggérant que le récepteur cardiaque AT1 serait une cible pour l'aldostérone [59].
6) Oestradiol
Le 17b-oestradiol est le plus puissant et le plus important des oestrogènes. Sa source principale est la conversion extraglandulaire de l'androstènedione, au niveau des tissus périphériques.
Les oestrogènes permettent le développement des caractères sexuels secondaires chez la femme, la croissance utérine, l'épaississement de la muqueuse vaginale, la fluidification du mucus cervical et le développement du système canalaire des seins.
Toutes les hormones stéroïdiennes sont dérivées du cholestérol provenant de trois sources : les lipoprotéines circulantes (surtout les LDL), le cholestérol synthétisé de novo dans l'ovaire et le cholestérol libéré des esters stockés dans les gouttelettes lipidiques.
La conversion du cholestérol en prégnénolone est l'étape limitante de la stéroïdogenèse ovarienne, qui à lieu au niveau des cellules de la granulosa, des cellules thécales et surtout au niveau du corps jaune.
Pour être transporté dans le sang, l'oestradiol est lié à l'albumine à 60%, à la globuline liant la testostérone à 38% et 2-3% est libre.
Les hormones stéroïdiennes ont un faible poids moléculaire et pénètrent facilement dans la cellule en diffusant à travers la membrane, bien qu'il puisse y avoir un transporteur qui participe à ce transport ; elles entrent dans le noyau et se lient à un récepteur nucléaire. L'affinité, la spécificité et la grande concentration des récepteurs aux stéroïdes permettent à une petite quantité d'hormone de produire un effet biologique. La spécificité des récepteurs aux oestrogènes est partiellement déterminée par le nombre de récepteurs dans la cellule. Par exemple, dans l'utérus, l'action biologique des oestrogènes est grande grâce à la concentration élevée des récepteurs dans une cellule. Il se produit une transformation active du complexe de l'hormone-récepteur après la liaison de l'hormone au noyau. Cette liaison implique un changement de la conformation qui, soit rend accessible des sites DNA, qu'on appelle steroid response elements (SREs), soit altère des complexes de la régulation de la transcription, pour leur permettre d'interagir avec des SREs pour activer ou réprimer des gènes [60].
Aucune étude n'a été publiée sur l'effet de l'oestradiol sur l'expression de la COX-2 dans les cardiomyocytes. Mais nous savons qu'il existe sur les cardiomyocytes des récepteurs aux oestrogènes.
En effet, la différence qu'il existe dans les maladies cardiovasculaires entre les deux sexes a suggéré que les oestrogènes agissaient directement sur le tissu cardiaque. C'est pourquoi Grohe et al. (1997) [61] ont cherché à savoir si les cardiomyocytes et les fibroblastes possédaient des récepteurs aux oestrogènes. Ils ont découvert par une méthodes d'immunofluorescence que le récepteur protéique aux oestrogènes était exprimé dans les cardiomyocytes et les fibroblastes cardiaques. La translocation nucléaire du récepteur protéique aux oestrogènes a été démontrée après stimulation des cardiomyocytes avec du 17-b-oestradiol (E2) [61].
Dans d'autres cellules que les cellules cardiaques, par exemple, les cellules musculaires lisses vasculaires, l'E2 stimule l'activité de la COX-2 et de la PGI2 synthase [62].
Ghanam K. et al. (2000) [63] ont vu avec l'aorte du lapin hypercholestérolémique que l'E2 restore l'effet relaxant de l'Acétylcholine (Ach) qui était inhibé par l'hypercholestérolémie.
Dans l'endomètre également, on a constaté que le pic de l'expression de la COX-2, exprimé durant les trois phases du cycle menstruel, correspond à la phase diestrus, qui correspond au niveau maximal d'E2, ce qui suggère une induction de la COX-2 dépendant de l'E2 [64].













