1. Introduction
Les néoplasies sont une complication fréquente de tout état d'immunodéficience.
Le syndrome d'immunodéficience acquise (SIDA), en tant qu'état d'immunodéficience acquise, ne fait pas exception. Les néoplasies et en particulier les lymphomes non-hodgkiniens (NHL) représentent une complication courante mais tardive d'une infection par le virus de l'immunodéficience humaine (HIV).
La lymphogénèse est un processus à multiples étapes que partiellement comprises. Ces différentes phases, chez les patients HIV+, se développent rapidement en comparaison à la population immunocompétente (moins de 6ans contre 30-40 ans).
Chez l'enfant, la survenue d'un cancer est moins fréquente que chez l'adulte. Il s'agit alors en majorité de NHL et de leïomyosarcome. Ces derniers ne sont cependant pas considérés comme un facteur définissant un SIDA. Il existe, par ailleurs, des cas de sarcome de Kaposi (KS), d'hépatoblastome et de leucémie lymphoïde aiguë à cellules B (B-LLA).
L'incidence des néoplasies chez les sujets HIV+ augmente avec la sévérité et la durée de l'immunodéficience.
Les NHL sont fréquemment l'événement à l'origine de la découverte d'une séropositivité ou celui inaugurant un SIDA.
Les NHL se développent chez toutes les catégories de patients HIV+, à tout stade de l'infection, dans des sites anatomiques variés et parfois inhabituels. Une localisation au niveau du système nerveux central (SNC) ou du tractus gastro-intestinal est particulièrement fréquente.
Les NHL rencontrer chez les patients HIV+ ont certaines caractéristiques en commun comme leur localisation préférentiellement extra ganglionnaire, leur haut grade de malignité, leur agressivité productive d'un stade avancé (III-IV) et leur histologie de type B.
La répartition des NHL en sous-type histologique est difficile compte tenu de la complexité de la classification des cellules du système lymphoïde et des différences d'interprétation des pathologues.
La présentation clinique des NHL et leur risque évolutif sont spécifiques du type histologique d'où l'importance d'un diagnostic de certitude.
L'agressivité de ces lymphomes est bien connue et l'évolution est, le plus souvent, rapidement défavorable.
Le traitement des NHL, une polychimiothérapie en priorité, chez les patients HIV+, doit être adapté en fonction de divers paramètres. Ces derniers sont en rapport avec le pronostic du NHL, lui-même dépendant de l'état d'immunosuppression du patient. Il s'agit principalement du compte de CD4 et de l'existence ou non du diagnostic de SIDA antérieur à celui du NHL
Un traitement curatif ou prophylactique du SNC ainsi qu'une prévention de la pneumocystose et un traitement anti-viral doivent êtres systématiquement instaurés.
La réponse aux différents protocoles de chimiothérapies n'est pas aussi bonne que dans la population générale et les rechutes sont précoces. Les décès sont soit secondaires à la progression de la tumeur soit aux infections opportunistes compliquant la chimiothérapie. La médiane de survie globale est inférieure à un an.
À l'image du cas étudié dans ce travail, on retrouve cependant dans la littérature de rares cas qui ont un meilleur pronostic.
L'évolution peut ainsi être favorable dans certains cas, si un traitement adéquat est entrepris précocement.
Existe-il certains facteurs qui permettent de prédire le mode d'évolution ?
Parmi ces facteurs, en existe-il qui sont génétiquement déterminés ?
Quelle est la fiabilité des éventuels facteurs pronostic ?
Quelle est la qualité de vie des patients HIV+, le jour où ils se retrouvent en rémission complète, compte tenu de la pathologie sous-jacente persistante, le SIDA ?
Voici quelques questions auxquelles nous allons tenter de répondre dans ce travail.
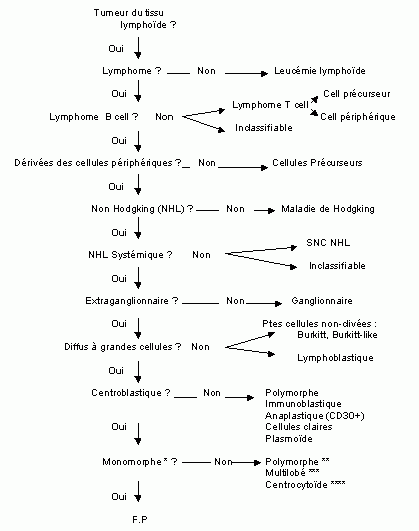
1.1 Classification des lymphomes
On regroupe sous le terme de " lymphome " une variété de proliférations malignes du système lymphoïde.
Compte tenu de la complexité du système immun, il n'est pas surprenant que les tumeurs dérivées de celui-ci - ainsi que leurs classifications - soient nombreuses et complexes(01). (voir tableau I)
Un diagnostic de certitude est possible, mais nécessite une description morphologique et histologique précise (taille et forme -clivé ou non- des noyaux cellulaires et le mode de croissance cellulaire -diffus versus folliculaire-)(02) ainsi que des analyses multiples telles que le réarrangement de gène(s), la cytochimie, l'immunophénotypie et la cytogénétique..)(01).
Une distinction est faite parmi les lymphomes selon leur histologie et leur localisation.
1.1.1 Distinction selon l'histologie du lymphome
-
Lymphome à cellules de type T ou lymphoblastes à l'origine des :
- Lymphoblastosarcomes
-
Lymphome à cellules de type B à l'origine des :
- Lymphomes de Hodgkin type I, II, III et IV
- Lymphome Non-Hodgkinien (NHL) : La majorité des NHL systémiques et du système nerveux central (SNC).
-
Lymphome de type histologique Indéfini
- Tous les " body cavity-based lymphoma " (BCBL) et la majorité des lymphomes anaplastiques à larges cellules type Ki-1 (ALC Ki-1+) n'expriment ni antigène cellulaire B ni antigène cellulaire T. Ceux-ci représentent 3 % des lymphomes(03).
1.1.2 Distinction selon la localisation du lymphome
-
Les lymphomes systémiques
- À localisation nodale.
- À localisation extra nodale (touchant préférentiellement le système nerveux central, le tractus gastro-intestinal et potentiellement tous les organes).
- Les " body cavity-based lymphoma " : Ils touchent exclusivement les cavités et y restent localisés. Ils se manifestent sous forme d'effusion lymphomateuse limitée à une cavité.
En 1966 Rappaport établi une première classification qui distingue quatre groupes de lymphomes :
- Les lymphomes lymphocytaires bien différenciés ou peu différenciés.
- Les formes mixtes (lymphocytaire & histiocytaire)
- Les formes histiocytaires.
- Les types indifférenciés.
Entre les années 1974 à 1982 plusieurs classifications sont décrites dont celles de Dorfman, de Kiel et de Lukes Collins
La classification de Kiel (dite de Lennert) est basée sur le potentiel malin des lymphomes.
La classification de Lukes-Collins repose sur des critères morphologiques et immunologiques.
En 1981 est développée la formulation internationale à usage clinique (Working Formulation) qui définit quatre groupes de NHL sans faire de distinction entre les lymphomes de type cellulaire B et ceux de type cellulaire T(04). Il s'agit :
- Des NHL de faibles malignités comprenant les leucémies lymphoïdes chroniques (LLC), les lymphomes lymphoplasmocytoïdes, les lymphomes folliculaires à petites cellules, les lymphomes folliculaires mixtes à petites et grandes cellules).
- Des NHL de malignités intermédiaires incluant les lymphomes folliculaires à grandes cellules, les lymphomes diffus à petites cellules clivées, les lymphomes diffus mixtes à petites et grandes cellules et les lymphomes diffus à grandes cellules clivées ou non.
- Des NHL de malignité élevée dont font partie les lymphomes à grandes cellules immunoblastiques (formes plasmocytoïdes, à cellules claires, polymorphe et à composante épithéloïde), les lymphomes lymphoblastiques et les lymphomes à petites cellules non clivées Burkitt ou Burkitt-like.
- D'une classe regroupant diverses néoplasies comprenant les formes composites, mycosis fongoïde, histiocytique, plasmocytome extramédullaire et inclassable.
Cependant, les NHL sont souvent seulement répartis en trois - voir deux - catégories : Les NHL de bas grade et ceux de grade intermédiaire et haut. Chacune de ces catégories regroupe cependant des entités très diverses(05).
La classification dite de Kiel modifié décrite en 1988 classe les NHL selon leur malignité et leur type cellulaire (B ou T). On retrouve les NHL de faibles malignités et ceux de hautes malignités ainsi qu'un groupe de NHL nommé " formes rares ". En revanche, la classification de Kiel ne prend pas en compte les lymphomes extra-nodaux qui représentent 30 % de tous les lymphomes(04).
La classification REAL (Revised European American Lymphoma) définie durant le début des années nonante, deviendra en 1999 la classification de l'OMS.
Cette dernière distingue des entités clinico-pathologiques en fonction de leur morphologie, leur immunophénotype et leurs traits génétiques(04). Les particularités moléculaires telles que les aberrations chromosomiques, la présence de génome viral intégré, le réarrangement clonal des immunoglobulines (Ig) ou des récepteurs des cellules T (TCR) sont pris en compte(04). La prise en considération de tous ces paramètres doit désormais permettre d'obtenir des résultats reproductibles. Elle inclus toutes les néoplasies dérivées du tissu lymphoïde y compris la maladie de Hodgkin et les myélomes(04). La classification REAL est subdivisée en 3 principaux groupes (cellules T, cellules B et maladie de Hodgkin). Dans les deux premières catégories, une distinction est faite entre les tumeurs dérivées des précurseurs (correspondant aux lymphomes lymphoblastiques et aux leucémies lymphoblastiques) et celles dérivées des éléments périphériques (comprenant tous les autres lymphomes et leucémies y-compris les catégories " provisoires " et " inclassifiable " par manque de consensus les concernant ou d'informations incomplètes)(04).
À relever que contrairement aux précédentes classifications, la classification REAL ne tient pas compte du degré de malignité(04).
En ce qui nous concerne, nous nous référerons uniquement à la classification REAL établie au début des années nonante et devenue en 1999 la classification de l'OMS.
Cette classification devrait être préférée aux autres, en particulier la " Working Formulation " encore souvent utilisée dans de nombreuses études. La classification REAL permet d'identifier des entités distinctes avec un comportement distinct et devrait permettre un traitement plus approprié avec de meilleures chances de succès(05).
Voir tableau II
1.2 Cyto-histologie (06)
1.2.1 Répartition histologique
La répartition des formes cyto-histologiques diverge entre les séries en raison des différences d'interprétation des pathologues et des variations géographiques.
La répartition histopathologique diffère entre la population générale et les patients HIV+ ainsi qu'entre les différents types d'immunodépression(03).
Chez les patients HIV+, tout comme dans la population pédiatrique (HIV+ ou -), trois types cyto-histologiques rendent compte de plus de 90 % des NHL (voir tableau III) alors qu'ils ne représentent que le 10-20 % des lymphomes dans la population générale(01, 02, 03, 07, 08, 09, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20). Ce sont :
1.2.1.1 Les lymphomes de Burkitt, de type Burkitt et de haut grade de malignité à cellularité B
Ils sont souvent associés au HIV chez l'adulte (1000 fois plus fréquent que dans la population générale)(03, 21, 22) et représentent 30 à 50 % des lymphomes pédiatriques. Le lymphome non-Burkitt est rare chez l'enfant. L'incidence est de 1 à 3 /million aux USA, 50 à 100/million en Afrique avec une prédominance masculine (rapport homme/femme = 2-3/1)(01, 21). Chez l'adulte, l'agressivité des lymphomes Burkitt-like est importante et le pronostic souvent fatal.
En revanche, dans d'autre état d'immunosuppression, les lymphomes de Burkitt sont rares, la majorité des NHL comportant des lymphomes à grandes cellules et des lymphomes/leucémie à cellules précurseurs lymphoblastiques T et B(03).
Histologie : Presque toujours de type B(01, 10) et EBV + dans 25 % des cas (01, 21). La croissance cellulaire est rapide avec un temps de dédoublement estimé à 24 heures responsable à l'examen histologique d'un aspect dit "de ciel étoilé ". C'est une néoplasie agressive mais curable. La forme Burkitt-like semble être une entité borderline entre le lymphome à grandes cellules et le lymphome de Burkitt. Le réarrangement du gène c-myc est souvent absent mais le réarrangement du gène bcl-2 est présent.
1.2.1.2 Les lymphomes/leucémies à cellules précurseurs lymphoblastiques T et B
Ils représentent 30 à 40 % de tous les lymphomes rencontré chez les enfants avec un pic d'incidence durant la deuxième décade. On note une prédominance masculine (rapport homme/femme 2 à 2.5). Ils sont plus rares chez l'adulte.
C'est une forme de NHL agressive, de haut grade de malignité mais souvent curable chez les personnes HIV- (01, 21).
Histologie : Il s'agit d'un infiltrat diffus monotone(01) presque toujours de type T(10). L'activité mitotique peut être prédominante et un ciel étoilé peut être observé(01, 21). On retrouve un réarrangement du gène des chaînes lourdes des immunoglobulines et parfois du gène des chaînes légères. Un réarrangement du gène T-cell receptor (TCR) est rare.
Il existe des anomalies chromosomiques variables dont t(1 ;19) et t(9 :28) qui influence la pronostic de façon défavorable. La présence de plus de 50 chromosomes entraîne en revanche un pronostic favorable(04).
Lorsqu'il y a plus de 25 % de lymphoblastes dans la moelle osseuse, on parle de leucémie lymphoïde aiguë (LLA), mais le terme de lymphome/leucémie lymphoblastique semble plus approprié puisque ces 2 entités semblent représenter 2 spectres d'une même maladie.
1.2.1.3 Les lymphomes diffus à grandes cellules B
(03, 07, 08, 09, 10, 11) (en particulier immunoblastique et anaplastique) Ils représentant 15 à 25 % des lymphomes de l'enfant et 30-40 % des NHL chez l'adulte HIV- dont l'âge moyen est situé dans la 6ème décade(21).
C'est un groupe de néoplasies hétérogènes du point de vue morphologie et clinique comprenant des phénotypes de cellules T (21 %), B (75 %, dont un tiers n'ont pas d'immunoglobuline de surface) et nulle aussi appelée indifférenciée (4 %)(21, 23). Chez l'enfant l'incidence du type cellulaire T peut être plus élevée car la forme CD30+ est relativement fréquente(10, 21). Des phénotypes mixtes ont été décrits ainsi que des formes cellulaire B avec une réaction cellulaire T extensive, des formes de lymphomes à larges cellules B riche en petits lymphocytes T ou histiocytes (et inversement soit des formes T riche en cellules B) ressemblant à des lymphomes à cellule T ou encore des maladies de Hodgkin de type prédominance lymphocytaire(21).
30-40 % ont une translocation impliquant le gène bcl-6 (3q27) représentant probablement une forme de tumeur de novo très sensible à la chimiothérapie(04).
30 % montre un réarrangement du gène bcl-2 qui ont un comportement plus agressif et peut-être des néoplasies secondaires à des lymphomes à cellule " centre folliculaire "(04, 21). Le réarrangement du gène c-myc est rare(04, 21).
Les lymphomes diffus à grandes cellules ont une croissance rapide et sont agressifs mais potentiellement curables avec une chimiothérapie agressive.
Les lymphomes diffus à grandes cellules ont des apparences morphologiques distinctes(01). Leur sous-classification est difficile, seul 4 % des coupes histologiques mettant l'ensemble des pathologues unanimes(21).
Les sous-classes histologiques, selon la Working Formulation, sont les suivantes :
- Les lymphomes centroblastiques représentent 75 % des lymphomes diffus à large cellules B. Ils sont subdivisés en quatre sous-groupes qui sont les lymphomes monomorphes (13 %), les lymphomes polymorphes (45 %), les lymphomes multilobés (17 %) et les lymphomes centrocytoïdes (11 %). Ce dernier est considéré comme une variante agressive des lymphomes à cellule manteau et est associé à une haute incidence d'infiltration médullaire. Les 14 % restants sont soit définis comme des lymphomes centroblastiques secondaires (12 %) soit sont non typisés (2 %)(24). La survie globale ne varie pas entre ces sous-groupes(24).
- Les lymphomes à grandes cellules clivées (noyau irrégulier)
- Les lymphomes immunoblastiques sont le sous-type le plus fréquent chez l'enfant. Sa morphologie est variée. Ils possèdent un important cytoplasme basophile et un grand noyau solitaire ou multiple centré sur un nucléole(24).
- Les lymphomes plasmacytoïdes ont un noyau rond-ovale, excentré avec un grand nucléole central et un important cytoplasme.
- La variante à cellules claires à un noyau de forme variable et un cytoplasme clair. Souvent de type cellulaire T.
- La variante polymorphe dont l'anaplastique à large cellules (CD 30 +, Ki-1) qui ont pour caractéristique de réagir avec l'anticorps monoclonal Ki-1/BerH2 (CD30). Ce dernier est considéré comme marqueur des cellules de Reed-Sternberg lors de maladie de Hodgkin et est identifié sur les lymphocytes B et T activés (25). Ces cellules sont plus larges et irrégulières que les immunoblastes et ont un cytoplasme moins basophile. Le pic d'incidence est situé dans la 2ème décade de vie.
- Les lymphomes anaplastiques à grandes cellules sont sous-divisés en 4 sous-types histologiques (commun, " Hodgkin-related ", riche en cellules géantes et lympho-histiocytique) (25) et sont, dans de nombreux cas, de type cellulaire T(24).
Il existe d'autres rares sous-classes de lymphomes à larges cellules. Il s'agit :
- Des lymphomes folliculaires, fréquents chez l'adulte mais rare chez l'enfant où ils restent souvent localisés.
- Des lymphomes à cellules B primairement médiastinaux, dérivant des cellules B thymiques. Ils se rencontrent surtout chez la jeune femme et parfois chez l'enfant.
- Les lymphomes B multilobulés, les lymphomes T périphériques, les lymphomes T cutanés avec ou sans phénomène de Sezary, les lymphomes de grade bas ou intermédiaire dont les lymphomes diffus à petites cellules lymphocytiques, les lymphomes diffus mixtes à petites et grandes cellules(01).
Chez les patients séropositifs, on rencontre d'autres néoplasies hématopoïétiques dont la fréquence et le pronostic est similaire chez les sujets HIV+ et dans la population générale. Ces néoplasies ne sont pas admises par le CDC comme critère de diagnostic d'un Sida. Il s'agit des lymphomes B à bas grade de malignité, des leucémies lymphoïdes, des myélomes multiples, des plasmocytomes, des lymphomes anaplasiques à grande cellule CD30+, des leucémies myéloïdes aiguës...(03).
1.2.2 Caractéristiques histologique
La majorité (90 %) des lymphomes associés aux VIH est d'origine cellulaire B(03, 07, 11, 26, 27) à l'exception de rare cas de lymphome à cellule T, surtout lors d'atteinte cutanée(27).
Chez les enfants HIV+, il semble y avoir une répartition environ égale des immunophénotypes entre les 3 catégories histologiques (B, T et nulle)(10).
Les autres caractéristiques moléculaires rencontrées lors de NHL chez les sujets HIV+ sont : l'absence d'ADN du virus Ebstein-Barr (EBV), l'absence de réarrangement du gène c-myc et la polyclonalité(28).
Les lymphomes associés aux transplantations se distinguent par la présence constante du DNA de l'EBV(15, 28).
1.2.3 Mono- et poly-clonalité
Le caractère monoclonal ne semble pas constant.
Cependant, l'existence de lymphome polyclonal avec un clone majeur et des clones mineurs reste encore à prouver(03). Pour certains auteurs, jusqu'à 40 % des NHL liés au SIDA serait polyclonaux quelques soit l'histologie, alors que pour d'autres aucun lymphome n'est polyclonal(29). Il n'est pas défini si ces dissimilitudes sont réelles ou liées à une différence de sensibilité des méthodes.
En effet, l'absence de réarrangement clonal du gène de l'immunoglobuline n'exclut pas la monoclonalité du NHL. Lorsqu'il n'y a pas de réarrangement clonal du gène de la chaîne lourde de l'immunoglobuline, on peut trouver un réarrangement clonal de la chaîne légère de l'immunoglobuline, l'évidence d'une infection unique par l'EBV ou encore un seul allèle de réarrangement d'un proto-oncogène / gène suppresseur de tumeur (c-myc, p 53, bcl-6)(03). Le réarrangement du gène c-myc est exclusivement lié aux tumeurs monoclonales et non aux NHL polyclonaux(03, 15, 28).
Les lymphomes polyclonaux pourraient être la manifestation d'une dysrégulation de production des cytokines apparaissant précocement lors d'un SIDA comme c'est le cas avec les syndromes de lymphadénopathies généralisées. Ces lymphomes ressemblent aux lymphomes polyclonaux des transplantés.
Les lymphomes polyclonaux pourraient évoluer secondairement en lymphomes monoclonaux. Il existe, en effet, des patients chez qui une maladie monoclonale est retrouvée à certains sites anatomiques et une maladie polyclonale à d'autres sites (28).
D'autre part, la monoclonalité n'implique pas nécessairement la notion de cancer. En réponse à une stimulation antigénique, une expansion clonale peut se développer. Ces clones, chez les personnes immunosupprimées, y compris les HIV, peuvent échapper au contrôle de régulation clonale et devenir cliniquement apparents sous forme de tumeur. En cas de restauration d'une fonction immune adéquate, le lymphome peut se résoudre spontanément et disparaître(29).
1.2.4 Corrélation entre l'immunophénotype et le pronostic (10)
Les lymphomes à cellules B sont plus souvent localisés et ont un meilleur pronostic que les autres immunotypes de lymphomes.
L'expression de l'Ag CD30, retrouvé lors des lymphomes anaplasiques à grandes cellules CD30+ n'influence pas la survie.
1.2.5 Lien entre sous-types histologiques de NHL et stade de l'infection HIV (03, 06, 30)
Les NHL de type Burkitt ou de type Burkitt sont une manifestation précoce de l'infection à HIV (03, 19, 27, 29, 30, 31). Ils s'observent chez les patients jeunes, asymptomatiques, sans diagnostic de SIDA antérieur et avec un taux de CD4 élevé (>300/mm3).
Les lymphomes diffus à grandes cellules sont une manifestation tardive d'une séropositivité à HIV (19, 27, 29, 30, 31). Ils prédominent chez les patients âgés avec une forte déplétion lymphocytaire de type CD4.
Les " body cavity-based lymphoma " sont plus proches du point de vue évolutivité des lymphomes diffus à grandes cellules que des lymphomes de Burkitt ou de type Burkitt(03).
Il semble aussi, qu'un patient sans diagnostic de SIDA antérieur à celui de NHL, développe préférentiellement un NHL extra-nodal comparativement au patient sans diagnostic de SIDA antérieur qui développe d'avantage un NHL nodal(31).
1.2.6 Corrélation entre le sous-type histologique du lymphome, sa localisation et sa présentation clinique (01, 03, 06, 31) (tableau IV)
Une corrélation entre le type histologique d'un lymphome, sa localisation et sa présentation clinique existe tant dans la population HIV+ que la population HIV-, à tout âge.
Les lymphomes de Burkitt et Burkitt-like prédominent au niveau abdominal, ganglionnaire(31), médullaire et musculaire et ils diffusent rapidement(01, 03).
Les lymphomes diffus à grandes cellules B touchent des sites variés extra nodulaires mais préférentiellement le tube digestif, la cavité buccale et le cerveau. Ils restent longtemps localisés avant d'envahir un ganglion(01, 03, 31).
Les lymphomes/leucémies à cellules précurseurs lymphoblastiques B & T se présentent comme une masse médiastinale avec dissémination précoce au SNC, aux gonades ou à la moelle osseuse.
Les NHL du SNC sont surtout de type diffus à larges cellules.
Les lymphomes liés au SIDA présentent donc des différences biologiques, cliniques et comportementales selon leur localisation d'origine et leur histologie.
1.3 Pathogénie & Lymphogènèse
Le développement de lymphomes chez les patients HIV+ est en relation avec la déplétion lymphocytaire et est l'aboutissement d'une accumulation de multiples facteurs, tous pas encore complètement compris.
1.3.1 Rôle des virus
Les virus, dont certains herpes virus tels que le virus Ebstein-Barr (EBV), les virus des hépatites B et C, le human herpes virus 8 (HHV-8) ainsi que les papillomavirus (type 11, 16 et 32) sont associés à l'étiologie d'environ 20 % des néoplasies à travers le monde. Ces virus sont ainsi appelé " onco-virus ". Les papillomavirus sont fréquemment associés au carcinome anal et cervical, le HHV-8 au sarcome de Kaposi et aux " body cavity-based lymphoma "(32). Parmi ces néoplasies, 10 % sont liées à des rétrovirus dont HTLV-1, HIV et HHV-8 (KSHV).
Les rétrovirus sont responsables de cancers par des mécanismes directs (dysrégulation de la croissance cellulaire) et indirects (interaction avec les signaux cellulaires, induction de facteurs de croissance(33), perturbation de l'immunité de l'hôte). La diminution du répertoire des cellules T, la perte d'un contrôle immunologique des cellules transformées et de certains virus dont l'EBV favorisent l'émergence de néoplasies (33, 34).
Les tumeurs associées à des virus présentant des cibles immunologiques plus nombreuses que les tumeurs non associées à des virus, une thérapie immunologique pourrait être efficace(35).
1.3.1.1 HTLV-1 (Human T lymphoma virus 1)
C'est un virus à simple brin RNA, capable de s'intégrer dans le génome d'une cellule hôte comme un " pro-virus ". Cette intégration permet, en échappant au système immun, une infection durable à l'origine d'une néoplasie (lymphomes/leucémies à cellules T de l'adulte, cancers cervicaux invasifs, cancers à petites cellules du poumons, leucémie à cellule T chevelue HTLV1+...) après une longue période de latence. Cette transformation à lieu dans 1 cas/1000 porteurs et par an. Cela représente 2500-3000 cas/an dans le monde. Dans les zones d'endémie (Japon, Brésil, Caraïbes), le virus HTLV-1 est lié à plus de 50 % des néoplasies lymphoïdes de l'adulte. Le pic d'incidence se situe entre 40 et 60 ans.
La présentation clinique est variée allant d'une forme agressive à une forme chronique ou une forme dite " smoldering " ressemblant au mycosis fungoïde/syndrome de Sezary avec invasion cutanée.
Le produit du gène viral " tax " est responsable d'une augmentation de la transcription du gène cellulaire codant pour différents facteurs de croissance (dont la production par les cellules T d'interleukine IL-2 et de son récepteur IL-2R) à l'origine d'un phénomène autocrine responsable d'une prolifération polyclonale. Les gènes viraux sont aussi responsables de la dysrégulation de gènes cellulaires régulateurs tels que le gène p53. La transformation d'une cellule est suivie d'une expansion clonale dont dérive la tumeur. Ces néoplasies sont relativement résistantes aux chimiothérapies et ont un mauvais pronostic(33).
1.3.1.2 HIV et lymphome associé
Le risque de cancer est augmenté chez les personnes HIV+. Les néoplasies les plus souvent rencontrées sont les sarcomes de Kaposi et les NHL de haut grade(33).
L'infection par le HIV s'accompagne d'une stimulation des lymphocytes B activés chroniquement - non-spécifique du HIV car provoqué par de nombreux antigènes, mitogènes et autre virus dont l'EBV et le HIV lui-même - cliniquement manifeste sous forme une hyperplasie folliculaire (appelée syndrome de lymphadénopathies multiples persistantes) avec hypergammaglobulinémie, tous deux non-spécifique du HIV(03, 06, 11, 30). Seuls 20-40 % des immunoglobulines sont spécifiques du HIV(30).
HIV a un rôle dans l'initiation du lymphome en favorisant l'émergence de lymphome par l'immunosuppression qu'il provoque et par des aberrations immunologiques dont l'altération fonctionnelles quantitatives des CD4.Cette stimulation chronique peut prédisposer à la transformation des cellules B(30). Le premier " hit " pourrait être une erreur de réplication de l'ADN à l'origine d'une expansion clonale(30).
Cet état de stimulation chronique des lymphocytes B et l'augmentation des immunoglobulines sériques est décelable quelques semaines après l'infection par le HIV et coïncide avec la phase de virémie(30). L'hyperactivité persiste au-delà et peut être à l'origine, dans 20 % des ganglions lymphoïdes hyperplasiques, de l'émergence de clones de cellules B. Ces lymphocytes B, immortalisés mais non transformés(03, 30) (gène c-myc non réarrangé), représentent un état pré-malin et favorise l'émergence d'un lymphome mais n'y sont pas directement associés(03, 29, 30, 33, 36).
En effet, les adénopathies hyperplasiques précèdent l'apparition d'un lymphome chez seulement 30% des individus HIV+. Réciproquement, seul 5 à 10 % des personnes HIV+ avec des adénopathies hyperplasiques développeront un lymphome malin, dont la majorité est extra-nodale.
L'hypergammaglobulinémie est surtout de type IgG polyclonale mais également IgA et IgM chez les patients HIV+ avec un ARC 1 . On note également l'activation de phénomènes auto-immuns(13, 30) et dans 25 % des cas, la sécrétion de paraprotéïne mono et oligo-clonales(30). Ces paraprotéïnes peuvent démontrer une spécificité (parfois unique) pour divers antigènes du HIV, en particulier les gènes gag et pol.
Paradoxalement, malgré l'hyperactivité des cellules B il y a une diminution de la capacité à répondre aux néo-Ag et aux antigènes tels que la toxine du tétanos(30). Cette capacité restreinte peut-être attribuée, en partie au moins, au déficit en cellules T helper et au virus HIV lui-même. Il existe vraisemblablement une suppression du SAC (secondary antigen challenge) médiée par le HIV. Les patients HIV+, bénéficient ainsi de transfusion d'immunoglobuline.
Le virus HIV aurait d'une part une action indirecte par la production et la dysrégulation de cytokines(33) (dont IL-6 par les monocytes et les macrophages et IL-10 qui ont des fonctions autocrine et paracrine) à l'origine de la différentiation de cellules B(03, 20), et d'autre part une action directe sur la prolifération des cellules B et leur augmentation de sécrétion accentuée par les cellules T(30). Le mécanisme de stimulation reste, quant à lui, peu clair. Des mécanismes conventionnels (liaison Ag - récepteur cellulaire provoquant une expansion clonale et une différentiation des cellules B reconnaissant HIV) et non conventionnels (dont les mécanismes sont inconnus) sont invoqués.
Le génome du HIV est retrouvé dans les macrophages. L'insertion du LTR (long terminal repeat) du HIV provoque l'expression augmentée du c-fes oncogène(15)
Le génome du HIV n'est pas intégré dans les cellules B des NHL lié au SIDA(29, 32, 36), un rôle direct oncogénique du HIV semble peu probable(32).
Il existe aussi probablement un déficit intrinsèque aux cellules B suggéré par le manque de corrélation entre la diminution du nombre de CD4 et l'émergence d'un clone d'une part, et la non correction de ce trouble de l'immunorégulation par l'injection de cellules T helper non infectées(30). L'émergence d'un NHL de haut grade coïncide, chez le singe, à un état d'immunosuppression avancé(03).
1.3.1.3 Ebstein-Barr Virus (EBV) et lymphomes associés
L'EBV possède la capacité d'induire une infection latente des lymphocytes périphériques, de les stimuler et les faire proliférer chroniquement(20), les immortaliser et conduire ainsi au développement de lymphomes malins.
Il existe deux type de virus EBV.
- Le type A retrouvé dans 1/3 des maladies de Hodgkin et chez les patients HIV-.
- Le type B retrouvé lors des lymphomes de Burkitt endémique, des lymphomes non hodgkinien (NHL) lié au SIDA et d'autres lymphomes chez des personnes immunosupprimées ainsi que lors des lymphomes du nasopharynx.
L'EBV est impliqué dans l'étiologie des lymphomes de Burkitt endémique (Africain) (31) (voir tableau V) et dans la pathogénèse de nombreux désordres lympho-prolifératifs chez des personnes avec une immunodéficience congénitale, iatrogène ou acquise(03, 11). En revanche, l'EBV est rarement associé aux NHL chez les personnes HIV- (26).
L'incapacité de contrôler immunologiquement l'EBV est un facteur de risque majeur pour certains types de NHL, dont les lymphomes lymphoblastiques et pour la progression des ADP associées aux HIV vers certains types de lymphome(30, 32).
L'ADN de l'EBV est retrouvé dans 30-50 % des lymphomes systémiques associés à l'immunodéficience dont le SIDA (03, 13, 29, 30, 34) soit 30 % des lymphome diffus à grandes cellules B et des lymphomes de Burkitt, plus de 80 % des maladies de Hodgkin et des ALC-Ki-1+ (11, 26, 27) et dans env. 100 % des lymphomes immunoblastiques et des lymphomes du système nerveux central (03, 11, 14, 20, 26, 27, 29). Chez les patients HIV-, les lymphomes du SNC ne sont EBV + que dans 7 % des cas(18, 27, 35).
Puisque la majorité des lymphomes du SNC chez les personnes HIV+ est de type immunoblastique et que l'EBV est préférentiellement associé à ce type de lymphome, il reste à définir si la différence de contenu d'EBV entre lymphomes systémiques et lymphomes du SNC est fonction du site anatomique ou du type histologique(03).
Il semble donc y avoir une corrélation entre l'histologie d'un NHL et la présence ou l'absence de l'EBV(03, 29, 30, 32). Cependant, il reste à définir si la pathogénèse des différents types histologiques des NHL dépend d'agent spécifique distinct ou si les agents (virus) ont simplement un tropisme différent pour des cellules à différent état de différentiation(32).
Le rôle de l'EBV est suggéré dans la lymphogénèse, mais son rôle précis est controversé et peu clair(03, 31, 37, 38, 39). Chaque lymphome contient un type unique d'EBV ce qui tend à prouver que l'infection est antérieure à la lymphomatogénèse(03). L'infection par EBV induit une stimulation polyclonale chronique rendant les erreurs de réplication plus probable et donc l'altération génétique dont certaines translocations chromosomiques menant à la transformation complète(30). Cette transformation aura plus de chance de survenir dans une cellule infectée par l'EBV(13).
1.3.2 Rôle des cytokines
Certaines cytokines ont une fonction paracrine (IL-6) (03) ou autocrine (IL-10 stimule les cellules B et inhibe potentiellement les cellules T). L'HIV induit le relâchement de cytokines multiples et de facteurs de croissance dont certains (IL6, IL10) (20, 33, 34, 36) vont contribuer à l'activation, la différentiation et la prolifération chronique des cellules B expliquant ainsi leur hyperplasie(32).
Le taux sérique d'IL6 pourrait être un facteur prédictif de développement d'un lymphome malin et d'autres désordres lympho-prolifératifs chez les patients avec une infection à HIV symptomatique(03, 36). Le taux d'IL-6 semble plus élevé chez les patients HIV+ développant ultérieurement un lymphome (NHL) que chez les patients HIV+ ne développant pas de NHL et plus élevé chez les patients présentant des symptômes B que chez les personnes sans symptômes B(36, 40). En revanche, il n'y a pas de corrélation entre le taux d'IL-6 et le stade du lymphome. La corrélation entre le taux d'IL et le type histologique du NHL est incertaine(03).
Le taux d'IL-6 varie avec la réponse au traitement, les taux diminuant chez les patients obtenant une rémission et augmentant chez les patients se péjorant ou lors d'infection virale et bactérienne(36).
Il se pourrait que la diminution du taux d'IL-6 secondaire soit à l'usage de zalcitabine (seul ou en association avec une chimiothérapie) perturbe le mode de stimulation paracrine et provoque une régression de la tumeur(36).
Chez les patients avec un NHL, le taux d'IL-2, d'IL-2 récepteur et de récepteur à la transferrine est d'avantage augmenté lors de stade III-IV que lors de stade I-II(27).
1.3.3 Rôle des pro-oncogènes (bcl 1,2 et 6 - c-myc - ras) et des gènes suppresseurs de tumeur (p53, Rb)
Les proto-oncogènes (c-myc, ras), lorsqu'ils sont activés, peuvent provoquer des néoplasies en permettant une prolifération cellulaire infinie(29).
A l'opposé, les anti-oncogènes (p53, RB), lorsqu'ils sont altérés, entraînent provoquent une perte de contrôle de la division cellulaire avec, comme conséquence, une prolifération infinie.
Chaque réarrangement de gène est secondaire à des mutations particulières et spécifique d'un type de lymphome (03, 04, 20, 31, 32).
Le rôle de certains de ces gènes est controversé, ces altérations n'étant pas retrouvées dans tous les lymphomes(29, 30). Cependant, ces mutations entraînent un déficit de contrôle des lymphocytes B par les lymphocytes T.
(voir tableau VI).
Le gène c-myc situé sur le chromosome 8 est souvent transloqué sur un locus de l'immunoglobuline (le plus souvent le gène de la chaîne lourde), t(8 :14)(q24 :q32)
Il est réarrangé dans 80 % des NHL lié au SIDA, soit 100 % des lymphomes de Burkitt (14, 20, 33), 50 % des lymphomes à grandes cellules et 25 % des lymphomes immunoblastique B (sous classe de lymphomes B à grandes cellules)(03).
En revanche, les lymphomes primaires du SNC et les BCBL ne montrent pas de réarrangement du gène c-myc.
Les lymphomes diffus à larges cellules semblent liés à une expression excessive du gène bcl-6(33).
Contrairement au concept généralement reconnu selon lequel la genèse d'une tumeur est un processus à multiples étapes (conséquence de l'association d'événement distinct) apparaissant sur une longue période de temps (30-40ans), chez les sujets HIV+ l'accumulation de multiples lésions génétiques se fait sur une relative courte période entre l'infection par HIV et le diagnostique de NHL (4-6 ans) (03). Les mécanismes de la lymphogénèse sont nombreux et varient selon le type histologique et le site anatomique d'origine du lymphome(31). Certains mécanismes peuvent être spécifiques des patients HIV+ (03).
1.4 Un schéma hypothétique de la lymphogénèse est le suivant
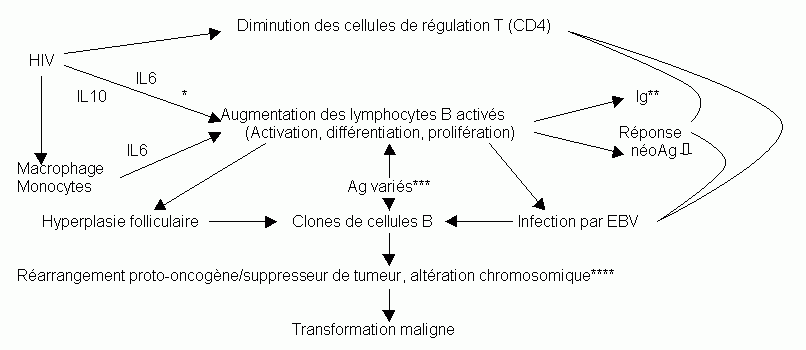
* : Stimulation conventionnelle ou non-conventionnelle
** : Hyper globulinémie
*** : Persistance d'Ag variés non éliminés chez les personnes immunodéficientes
**** : c-myc, ras, p53 et RB.
Risque fortement augmenté surtout dans les cellules EBV+. Prolifération cellulaire augmentée et diminution des corrections / éliminations cellulaire chez les personnes immunosupprimées(13).
L'apparition d'un lymphome est la conséquence de l'association d'événements distincts multiples.
L'infection par HIV cause une immunosuppression qui rend possible l'infection par l'EBV et la persistance d'antigènes variés à l'origine de l'activation polyclonal des cellules B(13). Cette dernière pourrait aussi être, en partie du moins, liée à un processus de compensation de l'organisme atteint par le HIV face à la diminution de ces capacités immunitaires(32). La prolifération des cellules B favorise les accidents génétiques tel qu'un réarrangement du gène c-myc. Apparaît alors un lymphome(32).
Lors d'infection par le HIV, le mécanisme de surveillance anti-tumoral est lui aussi défectif(39) permettant plus facilement la prolifération des cellules tumorales.
Le HIV est aussi à l'origine d'une sécrétion augmentée d'interleukines aux effets multiples (IL-1, IL-6, TNF).
Matériel et méthode (Casuistique)
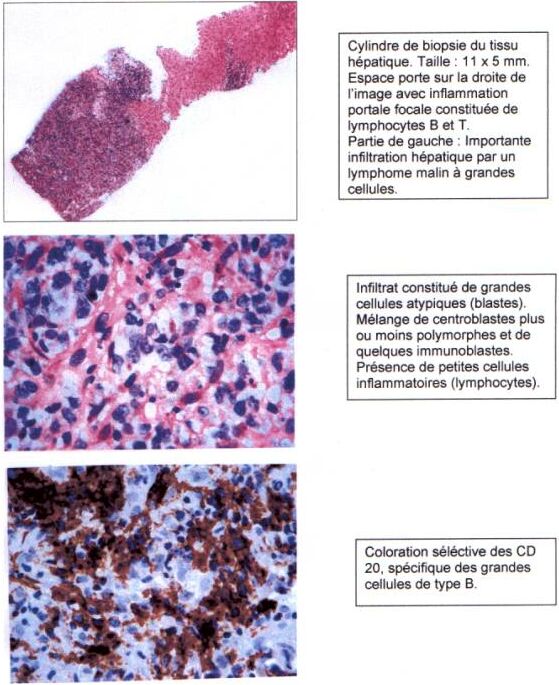
Il s'agit d'un cas de lymphome B hépatique à grandes cellules traité avec succès