2. Introduction
2.1 MHCII
Major histocompatibility complex (MHC) molecules, also called human leukocyte antigens (HLA) in human and Ia or H-2 antigens in mouse, were first detected as cell surface antigens that varied between different strains of mice and that were encoded by several genes responsible for the rejection of transplanted tissues (118). Later, the genes controlling immune responses in transplantation and specificity of antigen recognition by T cells were assigned to the same MHC loci (119-121). Since then, much has been learned about the structure, diversity and function of MHC molecules.
2.1.1 MHCI versus MHCII molecules
There are two classes of MHC molecules, class I (MHCI) and class II (MHCII). MHCI and MHCII molecules are both transmembrane glycoproteins belonging to the immunoglobulin supergene family, and their structures are very similar. However, their polypeptide chain composition, patterns of expression and functions in the immune system are very different (Fig.1).
MHCI molecules are composed of a transmembrane a chain associated non-covalently with the b2-microglobulin (b2m) chain. They are expressed on essentially all nucleated cell types. MHCI molecules are specialized for the presentation of peptides derived from endogenous proteins to the T cell receptor (TCR) of CD8+ T cells.
MHCII molecules are composed of two non-covalently linked transmembrane chains, called a and b. In contrast to the ubiquitous expression of MHCI, MHCII molecules are expressed only on a subset of cells. They are specialized for the presentation of extracellular antigens to the TCR of CD4+ T cells.

A. Interaction of a peptide-loaded MHCI molecule on any cell type with the TCR of a CD8+ T cell. B. Interaction of a peptide-loaded MHCII molecule on an antigen presenting cell (APC) with the TCR of a CD4+ T cell.
2.1.2 The role of MHCII molecules in the immune response
MHCII is one of the key molecules for the development of a specific immune response to a pathogen (8, 9). MHCII-peptide complexes displayed at the surface of antigen presenting cells (APCs) are recognized by the TCR and the CD4 co-receptor on CD4+ T cells. This triggers activation and proliferation of the T cells and thus elicits an immune response specific for the antigen from which the MHCII bound peptides were derived.
MHCII molecules are also crucial for selection and maturation of CD4+ T cells in the thymus. Positive selection, which ensures the survival of T cells that carry TCRs capable of recognizing self-MHC molecules, is driven by MHCII positive epithelial cells in the thymic cortex (cTECs) (23). On the other hand, elimination of autoreactive T cells by negative selection is driven by MHCII positive thymic dendritic cells in the medulla (122, 123).
2.1.3 The structure of MHCII molecules
MHCII molecules belong to the immunoglobulin gene family. They are heterodimeric glycoproteins consisting of two non-covalently linked transmembrane chains called a (33 kD) and b (29 kD). The difference in size of the two chains is mainly attributed to differences in N-linked glycosylation. The a and b chains have the same overall conformation, each consisting of two extracellular domains, a1 and a2, and b1 and b2, respectively. The membrane-distal domains combine to form a single peptide binding groove composed of two antiparallel a-helical loops supported by a platform of eight antiparallel b strands (124) (Fig.2). The groove is capable of binding a wide range of peptides. Peptides binding to MHCII molecules are at least 13 amino acids long and can be much longer. Their N- and C-termini may extend beyond the ends of the groove. The peptides are held in the peptide-binding groove both by peptide side chains (anchor positions) that protrude into polymorphic pockets lined by residues that vary between MHCII molecules, and by interactions between the peptide backbone and the side chains of residues that are conserved in all MHCII peptide-binding grooves.
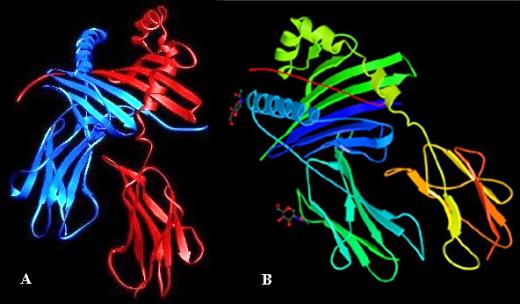
A. X-ray structure of HLA-DR1 with the a chain in blue and the b chain in red. B. Complex between HLA-DR3 and the CLIP peptide (in red). CLIP and antigenic peptides bind in an almost identical manner (125).
2.1.4 The diversity of MHCII genes and their genomic organization
Three classical MHCII molecules exist in man: HLA-DP (3), HLA-DQ (4) and HLA-DR (2, 5). Mice only express proteins orthologous to the last two, I-A (6) and I-E (7), respectively. In addition, both species encode so-called nonclassical molecules, namely HLA-DM (126) and HLA-DO (127, 128) in human, and H2-M (129) and H2-O (130) in mouse. The a and b chains of each MHCII molecule are encoded by separate genes in the class II region of the MHC (1) (Fig.3). The human MHC is located on the short arm of chromosome 6, while the mouse MHC is situated on chromosome 17. In all cases, except for HLA-DO, the pairs of genes are encoded adjacently. Remarkably, the class II region of the MHC is full of pseudogenes. These may be involved in generating new alleles by gene conversion. The class II region appears to have been subjected to several duplications generating novel gene family members, which have then diverged into new functions. The duplications must have taken place at several different periods throughout evolution of the class II gene family. HLA-DM, for instance, is only weakly related to other class II sequences, thus resulting from an ancient gene duplication, whereas HLA-DO shares about 60% identity with HLA-DR, thus representing a more recent duplication.
The class II region of the human and mouse MHC harbors a small number of genes involved in antigen presentation by MHCI. These encode the TAP transporter (TAP1, TAP2) as well as subunits of the immunoproteasome (LMP2, LMP7). In addition, many loci exist in the MHC that do not play any role in the immune system (131).
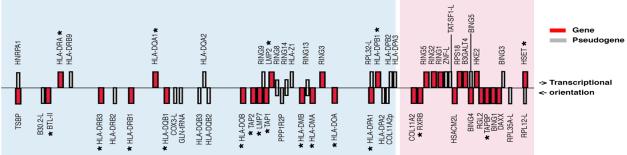
The class II region of the human MHC extends over 1'000 kb from its centromeric to telomeric end (from right to left) of the short arm of chromosome 6. The approximate positions and transcriptional orientations of all identified genes (in red) and pseudogenes (in gray) are shown. The expressed immune loci are marked with an asterix. The classical class II region is highlighted by a blue background, the extended class II region by a pink background.
2.1.5 Polymorphism
Classical MHCII genes exhibit an extraordinary degree of allelic polymorphism. There are more than 200 alleles of some MHCII loci. The polymorphic residues are primarily those that interact with the TCR and determine the shape of the peptide-binding groove. They consequently influence both the recognition of the MHCII-peptide complex by the TCR and the peptide specificities of different MHCII alleles. In addition, MHCII polymorphism contributes to the susceptibility to autoimmune diseases, has important functional implications in clinical organ and bone marrow transplantation, and provides a very useful set of markers for the field of human population genetics.
The nonclassical class II molecules are relatively invariant. Some alleles of both HLA-DM and HLA-DO have been described, but these vary by small numbers of amino acids and, so far, have no known functional significance. In addition, the human DRa chain has also been shown to be monomorphic.
2.1.6 Formation of MHCII-peptide complexes
MHCII molecules are synthesized in the endoplasmatic reticulum (ER). In the ER, three a/b MHCII dimers associate with a trimer of invariant chains (Ii) (10) (Fig.4). A number of functional consequences of the association of MHCII with Ii trimers are known. First, Ii serves as a scaffold to facilitate protein folding and assembly of MHCII molecules. Second, Ii occupies the class II peptide binding cleft, thereby blocking premature MHCII-peptide association. Third, Ii-association is critical for normal intracellular trafficking of MHCII molecules, since MHCII molecules synthesized in the absence of Ii aggregate and do not exit the ER (132, 133). The Ii-associated MHCII molecules exit the ER and are transported via the Golgi to the endocytic pathway, more precisely to a late endosomal antigen-processing compartment called MIIC (11). This process is regulated by two di-leucine endocytic signals present in the Ii cytosolic domain (134). Once in the MIIC, the Ii-associated MHCII complexes meet an acidic, protease rich environment, where the Ii chain is degraded by the stepwise action of aspartyl proteases and cathepsins (12, 13). This generates the intermediates Iip23 and Iip10 and finally CLIP (class II associated Ii peptide). CLIP is derived from Ii amino acid residues 81-104 and binds to the antigen-binding groove of MHCII molecules (Fig.2). MHCII molecules then become competent to bind antigenic peptides that come from the degradation of exogenous proteins. Efficient exchange of CLIP for antigenic peptides is catalyzed by HLA-DM. HLA-DM binds transiently to MHCII-CLIP and stabilizes an intermediate state where CLIP is released, allowing other peptides to bind (14-16). HLA-DM dependent peptide loading occurs within the context of other associated molecules, such as HLA-DO (135, 136) and tetraspanins (137). HLA-DO is a B-cell specific and pH-dependent modulator of HLA-DM. It effectively blocks HLA-DM function at the endosomal pH, while allowing HLA-DM action at the more acidic pH of the MIICs (135, 138, 139). In this way, HLA-DO action may skew the class II-peptide loading process toward acidic compartments. Once stable complexes of MHCII and antigenic peptides are formed, they are released from HLA-DM and transported to the cell surface.
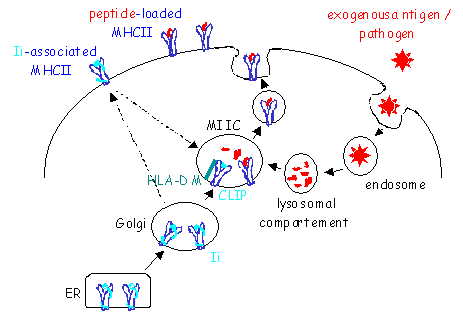
Newly synthesized ab dimers associate with Ii in the ER and are transported through the Golgi apparatus. The Ii-associated MHCII molecules are diverted from the constitutive seccretory pathway into the endocytic pathway. This can occur following a direct pathway from the Golgi to a specialized antigen-processing compartment (MIIC) or indirectly after transit through to the cell surface and endocytosis. In the MIIC, Ii is degraded leaving behind the CLIP peptide. Exogenous antigens are derived from proteins that are endocytosed and processed by proteases. The peptides bind to MHCII molecules replacing CLIP in an HLA-DM dependent manner, and the MHCII-peptide complexes are then transported to the plasma membrane.
2.1.7 Expression of MHCII molecules
The pattern of MHCII expression is complex and very tightly regulated. Under normal conditions, most classical and non-classical MHCII genes are regulated in a coordinated fashion. Two general modes of MHCII expression can be distinguished: constitutive and inducible (17-21).
Constitutive MHCII expression is largely restricted to APCs, such as dendritic cells (DCs), B cells and macrophages. In addition, certain epithelial and endothelial cells, most importantly cTECs, and activated human T cells express MHCII constitutively. Expression within an APC lineage can vary dramatically as a function of developmental stage. For instance, immature pre-B cells are MHCII negative, but become positive as they mature into B cells. Upon terminal differentiation of B cells into plasmocytes, MHCII expression is silenced again. In immature DCs, only few MHCII molecules are displayed at the cell surface, but they are upregulated dramatically with the induction of DC maturation. In many MHCII-negative cell types, MHCII expression can be induced by various stimuli of which IFN-g is the most potent and well known. Constitutive and inducible MHCII expression can be modulated by a large number of stimuli (17-21). For example, IL-4, IL-10 and IL-13 enhance MHCII expression in B cells, whereas glucocorticoids and prostaglandins diminish it. IFN-g induced MHCII expression is positively regulated by IL-4 and TNF-a, but negatively by TGF-b, IL-10, CSF-1, and type 1 interferons.
2.2 Regulation of MHCII expression
The molecular mechanisms regulating MHCII expression are the subject of a review that has been accepted for publication.
"CIITA and the MHCII enhanceosome in the regulation of MHCII expression"
Salomé Landmann, Jean-Marc Waldburger, Krzysztof Masternak, Annick Mühlethaler-Mottet and Walter Reith.
Current Genomics (2002), in press.
Summary
MHCII molecules direct the development, activation and homeostasis of CD4+ T cells. Given these key functions it is not surprising that the absence of MHCII expression results in a severe primary immunodeficiency disease called MHCII deficiency or the Bare Lymphocyte syndrome (BLS). The genetic defects responsible for BLS lie in genes encoding transcription factors required for MHCII expression. Four different MHCII regulatory genes encoding RFXANK, RFX5, RFXAP and CIITA have been identified. The first three are subunits of RFX, a ubiquitously expressed factor that binds cooperatively with other proteins to MHCII and related promoters to form a highly stable macromolecular nucleoprotein complex referred to as the MHCII enhanceosome. This enhanceosome serves as a landing pad for the MHCII transactivator CIITA. CIITA is a non-DNA binding coactivator that serves as the master control factor for MHCII expression. The highly regulated expression pattern of CIITA ultimately dictates the cells type specificity, induction and level of MHCII expression. The enhanceosome and CIITA collaborate in activating transcription by promoting histone hyperacetylation and by recruiting components of the general transcription machinery.
CIITA AND THE MHCII ENHANCEOSOME IN THE REGULATION OF MHCII EXPRESSION
S.Landmann, J.-M..Waldburger, K.Masternak, A.Muhlethaler-Mottet and W.Reith
Department of Genetics and Microbiology, University of Geneva Medical School
Abstract
Major histocompatibility complex class II (MHCII) molecules direct the development, activation and homeostasis of CD4+ T cells. Given these key functions it is not surprising that the absence of MHCII expression results in a severe primary immunodeficiency disease called MHCII deficiency or the Bare Lymphocyte Syndrome (BLS). The genetic defects responsible for BLS lie in genes encoding transcription factors required for MHCII expression. Four different MHCII regulatory genes encoding RFXANK, RFX5, RFXAP and CIITA have been identified. The first three are subunits of RFX, a ubiquitously expressed factor that binds cooperatively with other proteins to MHCII and related promoters to form a highly stable macromolecular nucleoprotein complex referred to as the MHCII enhanceosome. This enhanceosome serves as a landing pad for the MHCII transactivator CIITA. CIITA is a non-DNA binding coactivator that serves as the master control factor for MHCII expression. The highly regulated expression pattern of CIITA ultimately dictates the cell type specificity, induction and level of MHCII expression. The enhanceosome and CIITA collaborate in activating transcription by promoting histone hyperacetylation and by recruiting components of the general transcription machinery. In this review we summarize what is known about the molecular basis of BLS and what this has taught us about the mechanisms regulating transcription of MHCII and related genes. Particular attention is devoted to the structure, function and mode of action of the MHCII enhanceosome and CIITA. In addition, we focus on the highly regulated and cell type specific expression of CIITA.
Introduction
MHCII molecules are heterodimeric (a chain - b chain) transmembrane glycoproteins displayed at the surface of specialized cells of the immune system. In humans, there are three MHCII isotypes designated HLA-DR, HLA-DQ and HLA-DP. All three isotypes serve the same function, namely the presentation of peptides to the TCR of CD4+ T helper cells. This is crucial for numerous aspects of the adaptive immune system, including the selection, activation and survival of CD4+ T cells [1,2]. Two general modes of MHCII expression exist: constitutive and inducible [3-9]. Constitutive expression is the hallmark of professional APCs. These include B lymphocytes, cells of the monocyte/macrophage lineage and DCs. cTECs and activated human T cells are also MHCII positive. Most other cell types do usually not express MHCII molecules but can be induced to do so in response to various stimuli of which IFN-g is the most potent and well known. Both constitutive and induced MHCII expression can be further modulated by additional signals. Constitutive expression in B cells and DCs is for instance regulated as a function of developmental stage. MHCII expression is extinguished upon differentiation of B cells into plasmocytes. In DCs, maturation is accompanied by an increase in cell surface MHCII expression. Finally, IFN-g induced expression can be modified by various stimuli. For example TGF-b, IFN-a and IL-4 inhibit induction of MHCII by IFN-g.
The central importance of correctly regulated MHCII expression is underlined by the fact that deregulation of this expression leads to disease. Aberrant or inappropriate MHCII expression has been incriminated in the pathology of certain CD4+ T cell-mediated autoimmune diseases [10]. On the other hand, the lack of MHCII expression severely cripples the immune system and leads to a life-threatening immunodeficiency syndrome [7-9,11-14].A detailed understanding of the molecular mechanisms that control MHCII expression thus represents an important contribution to both molecular immunology and immunopathology.
The Bare Lymphocyte Syndrome
A disease resulting from the absence of MHCII expression
The absence of MHCII expression is the cause of a primary immunodeficiency syndrome called the Bare Lymphocyte Syndrome (BLS) [7-9,11-16]. BLS is a rare autosomal recessive disease and has a high incidence of consanguinity [17]. Since its first description in the late 1970s and early 1980s [18-23], only about 70 patients from 50 unrelated families have been reported.
Detailed descriptions of the clinical and immunopathological characteristics of the disease have been published previously [11-13,24], and we will thus restrict ourselves here to a brief description of the most salient features. Symptoms start within the first year of life, and comprise primarily severe and repeated infections, protracted diarrhea, malabsorption, and failure to thrive. The patients are particularly prone to infections of the gastrointestinal, pulmonary, upper respiratory and urinary tracts. The type of infectious agent is not pathognomic, and they can be of viral, bacterial, fungal or protozoan origin. Few children reach puberty. The majority dies between the age of 6 months and 5 years.
The mainstay of diagnosis is the absence of constitutive and inducible expression of MHCII genes [11-14,24]. Moreover, expression of the HLA-DM genes is suppressed and expression of the invariant chain Ii is reduced [25,26]. The level of MHCI expression is also reduced to a variable extent in many BLS patients. However, the clinical and immunopathological manifestations of the disease are thought to result mainly from the defect in MHCII expression.
Total numbers of circulating T and B lymphocytes are normal. However, in the majority of patients, the ratio of CD4+ to CD8+ T cells is reduced [11-13,24]. There is a reduction in the absolute number of CD4+ T cells and a concomitant increase in CD8+ T cells. This presumably reflects a deficiency in positive selection of CD4+ thymocytes, and is a consequence of the lack of MHCII expression on cTECs. Albeit reduced in numbers, the remaining CD4+ T cells in the patients do not exhibit major phenotypic abnormalities [27]. Although the residual CD4+ T cells in BLS patients may be functional, the absence of MHCII on APCs and the resulting inability to present antigens to CD4+ T cells leads to a severely compromised immune system [11-13,24]. Humoral immune responses to immunizations and to infectious agents are absent or strongly reduced. Cellular immune responses are also defective.
In all patients, the lack of cell surface MHCII expression is a consequence of the fact that the corresponding genes are not transcribed [28]. Several lines of evidence have shown that the defects responsible for the disease do not reside in the MHCII locus itself, but lie in transacting regulatory genes controlling MHCII expression [12,17,29-34] (Fig.5).
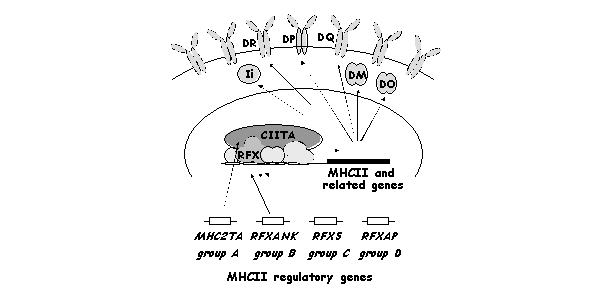
The MHC2TA, RFXANK, RFX5 and RFXAP genes are mutated in the BLS complementation groups A, B, C and D, respectively. These four genes are essential for the expression of MHCII and related genes. These include the genes coding for HLA-DR, HLA-DP, HLA-DQ, HLA-DM, HLA-DO and the invariant chain Ii.
Genetic heterogeneity in the cause of BLS
BLS is considered to be a single phenotypic entity. However, the disease is genetically heterogeneous [17,30-32]. Four different BLS complementation groups (A to D), reflecting the existence of mutations in four distinct MHCII regulatory genes, have been defined [17,30-32]. The regulatory gene affected in each complementation group has now been identified. The four affected genes are called MHC2TA (group A), RFXANK (group B), RFX5 (group C) and RFXAP (group D) [35-39]. The patient-derived cell lines, together with several experimentally generated MHCII negative mutants, have thus allowed the isolation and characterization of four key MHCII specific transcription factors and have hence constituted a unique tool for the investigation of the regulation of MHCII gene expression.
The nature of the genetic defect does not correlate with the course of the disease. There is no obvious genotype-phenotype relationship between the regulatory gene that is affected and either the immunological features, the precise clinical picture, the severity of the disease or the prognosis. Despite the genetic heterogeneity, BLS is therefore clinically homogenous.
Certain BLS patients display only mild symptoms or are even assymptomatic [40]. A patient exhibiting a very late onset of the disease (in his late twenties) has also been described [41]. Interestingly, in contrast to the situation observed in the majority of classical patients, the mutations identified in late onset patients do not destabilize the affected factor [40]. As the other 'classical' patients, these individuals completely lack MHCII expression or display only very low residual levels of MHCII molecules at the surface of their cells. Moreover, they have been assigned to one of the four known complementation groups. It is thus clear, that some patients display an attenuated clinical phenotype despite their profound defect in MHCII expression. This has two important implications. First, there are likely to be additional - as yet undefined - 'modifier' loci that have a major impact on the severity and course of the disease. Second, the frequency of the inherited MHCII deficiency disease is likely to be underestimated.
Lessons from mouse models of BLS
There are no spontaneous animal models for BLS. However, identification of the genes affected in the human disease has permitted the generation of two mouse models. Knockout mice reproducing the molecular defects of BLS patients in groups A and C have been constructed by deletion of the Mhc2ta and Rfx5 genes [42-45]. The major immunopathological characteristics of the human disease are reproduced in these mice. Both models show a strong reduction of constitutive MHCII expression on professional APCs (B cells, DCs and macrophages). IFN-g induced expression is also abolished. As in humans, this loss of MHCII expression results in a severely compromised immune system.
The absence of CD4+ T cell-dependent immune responses in the mouse models is a consequence both of the inability to present antigen via MHCII molecules and of a severe CD4+ T cell deficiency. The CD4+ T cell population in Mhc2ta and Rfx5 knockout mice is decreased over tenfold [42-45]. This strong reduction is a consequence of severely impaired positive selection, which results from an essentially complete loss of MHCII expression on epithelial cells in the thymus. Surprisingly, the reduction in CD4+ T cells in BLS patients is rarely greater than two to three fold [11,12] and is thus considerably less pronounced than in the knockout mice. At least two explanations could account for this difference between the human and mouse phenotypes. First, the relatively mild decrease in peripheral CD4+ T cells in BLS patients suggests that positive selection may be compromised only partially. This could be explained by the retention of a sufficient level of residual MHCII expression in the thymus. Whether or not this is indeed the case remains to be determined because MHCII expression patterns in the thymus have only been examined in a few isolated cases [46]. It may be relevant that residual expression of MHCII in the thymus has been observed in one patient [46]. A second interpretation could be that the CD4+ T cells in BLS patients have escaped the normal selection processes in the thymus. They may for example have been selected on ligands other than MHCII molecules. In MHCII deficient mice, for instance, a large proportion of the residual CD4+ cells are CD1-restricted [47]. Such alternative selection pathways could be more prominent in BLS patients. Interestingly, an analysis of the T cell repertoire in BLS patients has revealed minor alterations, suggesting that the CD4+ T cells in these patients may indeed have been subjected to an unusual selection mechanism [48,49].
Both the Mhc2ta-/- and Rfx5-/- mice exhibit residual MHCII expression in certain tissues and cell types. The precise pattern of residual expression differs between the two mice. This implies that specific cellular compartments possess alternative pathways for MHCII expression that can partially bypass the strict requirement for RFX5 and/or CIITA [42-44]. Rfx5-/- mice retain relatively strong MHCII expression in the thymic medulla and significant, albeit weaker, expression on a fraction of splenic and bone marrow derived dendritic cells. Low MHCII expression is also induced on B cells from Rfx5-/- mice following activation in vitro with lipopolysaccharide (LPS) and/or IL-4. In contrast, residual MHCII expression in the Mhc2ta-/- mice is mainly restricted to DCs in the paracortex of lymph nodes, B cells in the germinal centers of the spleen and lymph nodes, and a subset of cTECs. This difference in the residual expression pattern is surprising because the human disease is considered to be phenotypically homogeneous. Leaky expression has been observed in cells from certain BLS patients, but no characteristic residual expression pattern distinguishing RFX5 deficient patients from those with defects in CIITA have been described [11,12,24]. This discrepancy may reflect species-specific differences in the dependence on the two MHCII regulatory genes. However, it is also possible that the phenotypic differences observed in the mouse system exist in the human disease as well, but have escaped attention until now. Due to the rarity and severity of the disease, only relatively few patients from defined complementation groups have been studied in detail with respect to residual MHCII expression.
Therapeutic strategies
Carrier detection and prenatal diagnosis for BLS
Considering the rarity of BLS, prenatal/postnatal diagnosis or carrier detection on a population-wide scale is not justified. On the other hand, these procedures could be valuable if restricted to families that already have affected children, or when a consanguineous union is envisaged in a high risk population. Previously, prenatal diagnosis relied on the analysis of MHCII expression on fetal leukocytes obtained by an umbilical vein puncture [50]. With our current knowledge of the molecular defects in BLS, mutated alleles of MHC2TA, RFXANK, RFX5 or RFXAP can now be screened for by using flanking polymorphic markers or by a direct search for known mutations. The greater sensitivity of these molecular techniques will allow the development of prenatal diagnosis by less invasive procedures, such as choriocentesis or amniocentesis. In addition, thanks to these techniques healthy carriers can now be identified rapidly and given accurate genetic counseling.
Gene therapy for BLS
Allogeneic bone marrow transplantation (BMT) is currently the only curative treatment available for BLS [51]. A cord blood transplantation from an HLA-identical sibling has also been successful in the case of one patient with an allogeneic BMT graft failure [52]. The overall success rate of BMT in this disease has been reported to be relatively poor as compared to other immunodeficiency syndromes. This does not appear to be a peculiarity resulting from the immunological phenotype of BLS. Instead, it is likely to be due largely to other problems, such as the fact that the disease is often diagnosed at a rather late age, when recurrent illness will have compromised the success of BMT [51,53]. Now that the affected genes have been identified, gene therapy becomes a potential alternative to BMT. Introduction of the wild type MHC2TA, RFXANK, RFX5 or RFXAP genes into hematopoietic stem cells (HSCs) of BLS patients in complementation groups A, B, C or D, respectively, would represent a logical therapeutic strategy. The Mhc2ta and Rfx5 knockout mice will be valuable for developing gene therapy for BLS.
Abnormal selection of CD4+ T cells, resulting from the absence of MHCII expression on thymic epithelial cells, is unlikely to represent an obstacle for gene therapy in BLS. The fact that classical BMT can cure BLS suggests that restoring MHCII expression on thymic epithelial cells is not essential. This is consistent with the fact that successful BMT does not require a functional thymus; the T cell pool can be reconstituted through peripheral expansion of passenger donor T cells present in the graft [54]. In this respect it is also worth mentioning that in a MHCI deficient mouse model, MHCI expression on bone marrow derived cells was found to be sufficient to compensate for the defect in thymic selection of CD8+ T cells [55].
One potential concern is that the therapeutic transgene could induce ectopic or non-physiological levels of MHCII expression, which could have deleterious consequences and result in tissue destruction or autoimmunity. This is unlikely to be a major problem in the case of RFX5, RFXANK and RFXAP, which are expressed ubiquitously in all cell types. The MHC2TA gene, on the other hand, is tightly regulated and it will be difficult to obtain a pattern of transgene expression that mimics that of the endogenous gene.
RFX and CIITA as targets for novel immunomodulators
MHCII expression is completely lost or severely reduced in BLS patients. This is also the case in most cell types in the two mouse models for the human disease. This suggests that CIITA, RFXANK, RFX5 and RFXAP are essential and that no bypass or alternative pathways can compensate efficiently for their absence. Moreover, no other major systems are as critically dependent on these regulatory factors. These features imply that inhibitors specific for CIITA, RFXANK, RFXAP and RFX5 should induce a highly selective downregulation of MHCII expression. CIITA, RFXANK, RFXAP and RFX5 may thus represent prime targets for novel immunomodulatory drugs having wide applications in situations such as organ transplantation and autoimmune diseases, where inhibition of MHCII expression might be desirable or beneficial.
Tumor immunotherapy.
Tumors frequently have reduced immunogenicity because they lack MHCII molecules. The immunogenicity and rejection of such tumors can be increased by the expression of MHCII alone or in combination with costimulatory molecules [56-58]. This has of course raised hopes that the introduction of CIITA to activate MHCII expression might enhance tumor immunogenicity and thus contribute to the success of tumor therapy. Experiments of this type have been performed [59], although with limited success so far.
Regulation of MHCII expression
Overview
MHCII expression is regulated primarily at the level of transcription [3-9,14]. The promoter proximal region of MHCII and related genes contains the conserved cis-regulatory elements referred to as W (or S), X, X2 and Y 'boxes' (Fig.6). These elements are highly conserved in their sequence, orientation, order and spacing relative to each other, and they function together as a single composite enhancer unit [3-9,14].

a) The promoter proximal regions of MHCII and related genes contain the conserved W, X, X2 and Y cis-regulatory elements. These elements are bound by the RFX, X2BP and NF-Y complexes and a putative W binding protein. These factors function together as a composite unit called the MHCII enhanceosome. The latter recruits the master regulator CIITA to the promoters. b) CIITA is mutated in BLS complementation group A. In the absence of CIITA, MHCII and related promoters are occupied by the enhanceosome but are not transcribed. c) In BLS complementation groups B, C and D one of the subunits of the RFX complex is mutated. In the absence of an intact RFX complex, the enhanceosome can not assemble, CIITA is not recruited, and MHCII and related promoters remain silent.
The X box is bound by the trimeric factor RFX, which is composed of RFX5, RFXANK and RFXAP. The X2 box is recognized by X2BP [60,61]. Finally, the trimeric complex NF-Y, composed of NF-YA, NF-YB and NF-YC, binds to the Y box [62]. Several proteins have been described to bind to the W box, but none has been definitely demonstrated to be the functionally relevant W box binding protein. All of the factors binding to the cis-regulatory elements are required for MHCII gene expression. However, they are ubiquitously expressed and fail to account for either constitutive or IFN-g inducible MHCII gene expression. Instead, they bind cooperatively to the promoter to form a landing pad for an additional factor that plays an essential regulatory role [63]. This factor is the class II transactivator CIITA [35]. The latter ultimately determines the cell type specificity, inducibility and expression level of MHCII genes. CIITA is now widely accepted as being the master regulator of MHCII expression [9,64].
The RFX complex
The RFX (regulatory factor X) complex is composed of three subunits RFXANK, RFX5 and RFXAP. These factors are defective in BLS complementation groups B, C and D, respectively (Fig.6C). The three subunits of the RFX complex are unrelated and share no sequence homology. The gene encoding the 75 kD RFX5 subunit was identified by means of a genetic complementation approach using a cell line derived from a group C patient [36]. RFX5 was the fifth member of the RFX family of DNA binding proteins to be identified [65]. All members of this family share a characteristic DNA binding domain (DBD) referred to as the RFX motif [65,66]. The three dimensional structure of the RFX motif of one family member (RFX1) has been solved and was found to belong to the winged helix subfamily of helix-turn-helix proteins [67]. The DBD of RFX5 lies near its N-terminus [68] (Fig.7C). Outside of the DBD, RFX5 contains no obvious functional motifs. There is a centrally placed proline rich region, of which the function remains unknown.
A biochemical approach was used to isolate the gene encoding the 36 kD RFXAP (RFX associated protein) subunit. This approach relied on affinity purification of the RFX complex and sequencing of peptides derived from its 36 kD subunit [37]. RFXAP contains an acidic domain, a glutamine rich segment and a basic region resembling a bipartite nuclear localization signal (NLS) [69,70] (Fig.7D).

a) The 1130 amino acid CIITA protein contains an N-terminal acidic domain (DE), an adjacent proline/serine/threonine rich (PST) region, a central GTP binding domain and a series of leucine rich repeats (LRR) at the C-terminus. At least three nuclear localization signals (NLS 1 to 3) are found in the protein. The N-terminus functions as a transcription activation domain and can interact with the indicated general transcription factors and coactivators. The central and C-terminal domains are required for recruitment of CIITA to the MHCII enhanceosome and function as a dominant negative mutant in the absence of the N-terminal activation domain. Domains believed to be involved in self-association of CIITA are indicated. b) The hallmark of the 269 amino acid RFXANK protein is a C-terminal ankyrin repeat domain. This domain is sufficient for complementation of cell lines from BLS complementation group B. The N-terminal part of the protein contains an acidic (DE) domain. c) The 616 amino acid RFX5 protein is the only subunit of the RFX complex containing a DNA binding domain (DBD). This DBD lies in an N-terminal region that is sufficient for assembly with RFXANK and RFXAP. For complementation of RFX5 defective cell lines, a central region containing a proline rich domain (P) is required in addition to the DBD. d) The C-terminus of RFXAP is sufficient for complementation of BLS cells lines in group D. Expression of HLA-DR only requires a short segment spanning the glutamine rich region (Q). Expression of HLA-DP and HLA-DQ required a larger C-terminal region. The acidic region (DE) and NLS-like sequence are not essential for the function of the protein.
RFXANK, the gene encoding the smallest 33 kD subunit of RFX was isolated by a biochemical approach similar to that used to isolate the RFXAP gene [38]. The gene was named RFXANK because it encodes a factor that contains a protein-protein interaction domain consisting of ankyrin repeats (Fig.7B). The same gene was isolated independently by another laboratory [39]. They called the encoded factor RFXB to indicate that this factor is mutated in BLS complementation group B. The N-terminal region of RFXANK is rich in acidic amino acids [39,69].
RFX5, RFXAP and RFXANK assemble to form the RFX complex that binds to the X box of MHCII promoters. The complex pre-assembles in solution before binding to its target site. The RFX complex can be faithfully reconstituted with the three recombinant proteins, indicating that the complex does not contain additional subunits [68-71]. However, the stoichiometry of the three subunits in the RFX complex remains to be determined. The absence of any one of the three subunits destroys the RFX complex and eliminates its ability to bind to DNA. This is illustrated by cell lines from BLS complementation groups B, C and D, which all lack RFX binding activity [28,72,73]. Experiments with recombinant RFX proteins have also shown that the individual subunits or combinations of two of the subunits are not capable of binding specifically to DNA in band shift assays [68-70]. RFX5 is the only RFX subunit containing a well defined DNA binding domain, although DNA-protein crosslink experiments have shown that all three subunits contact the DNA at the X box [74].
Specific domains that are necessary for complex formation and DNA binding activity have been identified in each of the RFX subunits (Fig.7B-D). In RFX5, the N-terminus (amino acids 39 to 194) comprising the DBD is sufficient for interaction with RFXAP, RFXANK and X box DNA [68,69]. In RFXAP, the 49 C terminal amino acids spanning the highly conserved glutamine rich region are sufficient for complex formation and binding to the X box [69,70]. Finally, the ankyrin repeat region of RFXANK (amino acids 84 to 269) meets all the requirements needed to form an efficient DNA bound complex with RFXAP and RFX5 [69] [our unpublished data]. The minimal domain of RFXANK allowing RFX complex formation and DNA binding is sufficient to restore MHCII expression in cell lines from RFXANK deficient BLS cells [69]. In the case of RFXAP, the minimal 49 amino acid C-terminal region is sufficient to restore HLA-DR expression in BLS cells lacking RFXAP [70]. However, a longer segment of RFXAP is required to restore expression of HLA-DQ and HLA-DP [70]. The domain of RFX5 mediating complex formation and DNA binding (amino acids 39 to 194), is not sufficient for complementation of RFX5 deficient cells [68]. An additional domain located between amino acids 410 to 515 is required [68]. This region has been shown to mediate cooperative DNA binding between RFX and NF-Y [68].
Outside of the essential domains defined above, several features within RFX appear to be dispensable. An acidic region and a putative nuclear localization signal present in RFXAP can be removed or mutated without affecting the ability of the protein to restore MHCII expression in cells from group D [69,70]. The N-terminal acidic region of RFXANK is not essential because it can be removed without eliminating the ability to complement cells from group B [69] [our unpublished data].
The MHCII enhanceosome
The trimeric RFX complex binds with relatively low affinity to the X box of MHCII promoters. However, this binding is greatly stabilized by synergistic interactions with two other factors binding to MHCII promoters, namely with the trimeric NF-Y complex [68,71,75] and the dimeric protein X2BP [61,76,77]. X2BP has recently been proposed to contain CREB [78]. The combination of these cooperative interactions results in stable occupation of MHCII promoters.
The quaternary complex composed of RFX, X2BP (CREB), NF-Y and MHCII promoter DNA is extremely stable with a half-life greatly exceeding that of any of the possible secondary or ternary complexes [77]. RFX plays a crucial role in promoting this cooperativity. This important role of the RFX complex is strongly emphasized by the analysis of promoter occupation in RFX deficient BLS cells (complementation groups B, C and D). MHCII promoters in these cell lines are unoccupied and lack the DNaseI hypersensitive sites typically observed in RFX positive cells [79-81].
The stable higher order nucleoprotein complex that assembles at MHCII promoters has been coined the 'MHCII enhanceosome' [63]. The enhanceosome can be isolated from cell extracts by promoter pull down assays, and has been shown to contain all of the known subunits of RFX, X2BP (CREB) and NF-Y [63]. It may also contain additional components but these remain to be characterized.
Formation of the enhanceosome requires the presence and proper arrangement of all cis-regulatory elements, that is to say the W, X, X2 and Y boxes. The spacing between the X and Y elements is highly conserved at approximately two helical turns [82-84]. Correct stereoalignment is essential for promoter activation, probably because the X and Y box binding factors need to be assembled on the same side of the DNA helix. It was also observed that the spacing between the W and X boxes is critical for promoter activation [83,84] [A.Muhlethaler-Mottet, unpublished results].
In addition to the crucial role of the MHCII enhanceosome in enhancing the stability and specificity of promoter occupation, it constitutes a platform onto which the transcriptional coactivator CIITA is recruited. CIITA has been directly implicated as a key protein mediating transcriptional activation of MHCII genes (see below). However, it has been recognized recently that the enhanceosome also contributes directly to the process of transcription activation independently of CIITA [85]. The relative contributions of the enhanceosome and CIITA to transcription activation depends on the MHCII promoter [85]. For instance, the enhanceosome has a dominant role in histone acetylation and recruitment of general transcription factors (GTF) at the DMB promoter. At the DRA promoter on the other hand the dominant role in these processes is played by CIITA. Promoters such as DPB depend equally on both the enhanceosome and CIITA. The example of the DMB promoter demonstrates that the enhanceosome is not merely a landing pad for CIITA, but can actually support CIITA independent events contributing directly to transcription initiation.
B cells display constitutively occupied MHCII promoters, even in the absence of CIITA (cells in group A) [80,81]. However, CIITA is clearly required for promoter occupancy in IFN-g induced or CIITA transfected fibroblasts [86-88]. It is not clear how CIITA induces promoter occupation in these cells. One possibility is that binding of CIITA favors assembly and/or stability of the enhanceosome by interacting with many of its individual components (see below). Alternatively, CIITA may facilitate in vivo occupation indirectly by changing accessibility of the promoter DNA.
The master regulator CIITA
The enhanceosome is essential but not sufficient for appropriate MHCII gene expression. This is demonstrated by the study of B cell lines from BLS patients in complementation group A. Enhanceosome assembly, promoter occupation and the presence of DNase hypersensitive sites are all normal at MHCII promoters in these cells (Fig.6B). Yet, the MHCII genes are not transcribed. The defect in this complementation group lies in the MHC2TA gene encoding the regulatory factor CIITA [35]. MHC2TA was isolated by the same approach employed for isolation of the RFX5 gene, namely by complementation cloning using the in vitro generated cell line RJ2.2.5 (complementation group A) [35].
CIITA expression is a nearly absolute prerequisite for both the constitutive or inducible expression of MHCII genes [35,42,89-94].The expression profile of CIITA is the major determinant dictating the tightly controlled pattern of MHCII expression [9,64]. The following findings emphasize this key regulatory function of CIITA. 1) A quantitative correlation between MHC2TA and MHCII expression levels has been revealed by the analysis of a large number of human and mouse tissues and cell lines [95]. In addition, an experimental setup using a tetracycline inducible system has shown that the level of MHC2TA expression directly determines the level of MHCII expression [95]. 2) Most cell types do not express MHC2TA and are thus MHCII negative. Transfection of these cells with a CIITA expression plasmid is generally sufficient to render them MHCII positive [89,90,96,97]. 3) IFN-g induces expression of the MHC2TA gene, and this mediates activation of MHCII genes [89,96]. 4) Induction of MHCII expression upon activation of human T cells is mediated by upregulation of MHC2TA expression [98] [our unpublished data]. 5) B cells loose MHCII expression during their differentiation into plasmocytes because the MHC2TA gene is repressed [90,99]. Exogenous expression of CIITA is sufficient to re-establish MHCII expression in plasmocytes [90,99]. 6) Silencing of MHCII expression in trophoblast cells is caused by inhibition of MHC2TA expression [94,100]. 7) Cytokines modulating IFN-g induced MHCII expression, such as TGF-b, IL-1, IL-4 and IL-10, do so by modulating MHC2TA expression [101-105]. 8) Pathogens such as cytomegalovirus, varicella-zoster virus, Mycobacterium bovis and Chlamydia can downregulate MHCII expression by interfering with transcription of the MHC2TA gene [106-109]. In summary, CIITA exerts a strict qualitative and quantitative control over MHCII expression and thus deserves the title of the master regulator of MHCII expression [9,64].
The control of CIITA over MHCII expression is achieved mainly via the level of transcription of the MHC2TA gene. However, a few examples of other levels of control are known. The inhibitory effect of IFN-b and TNF-a on IFN-g induced MHCII expression is exerted downstream of MHC2TA transcription [110-112]. The transactivating capacity of CIITA can be modulated by posttranslational modifications. protein kinase A (PKA) can inactivate CIITA by phosphorylating it at serine residues 834 and 1050 [113]. Phosphorylation of CIITA by PKA is thought to be involved in the inhibition of MHCII expression by prostaglandin E (PGE), since PKA is a downstream effector in the PGE/cAMP cascade.
A few examples of CIITA independent MHCII expression have been reported. Isotype specific MHCII expression has been observed in certain mutant human cell lines devoid of functional CIITA [114]. MHCII expression has also been detected in certain tissues of CIITA-/- mice, although the levels are significantly lower than in control animals [42,44]. For the moment, these cases of CIITA independent expression remain unresolved exceptions.
Regulation of MHC2TA expression
The MHC2TA gene is controlled by multiple promoters
The MHC2TA gene resides in a locus known as AIR-1 on chromosome 16 (16p13) [115]. Mhc2ta, the homologous mouse gene, is localized in a syntenic region on mouse chromosome 16. The genomic organization of the entire mouse gene has been determined. It consists of 19 exons spanning 42 kb of DNA [our unpublished data].
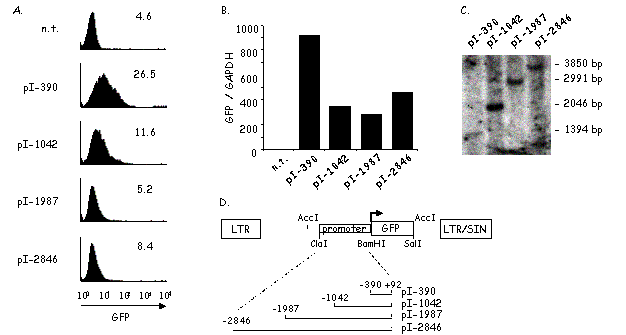
Expression of MHC2TA is controlled by four different promoters (pI to pIV) [116] (Fig.8). Three of these promoters are highly conserved between the human and mouse genes (pI, pIII and pIV). pII has only been found in the human gene. It displays only a very low transcriptional activity and its significance is not known. pII will thus not be discussed further. The different promoters do not share any sequence homology and are not co-regulated. They span a large (> 12 kb) genomic region. Each promoter precedes a distinct first exon that is spliced alternatively to a shared second exon. This leads to the production of three types of transcript (type I, type III and type IV), each possessing a different 5' end [116] (Fig.8).
The shared second exon contains an AUG translation initiation codon that can be used in all three types of transcript to give rise to a 1106 amino acid protein. However, the first exons of the type I and type III transcripts each contain an additional in-frame translation initiation codon. Usage of these alternative start codons leads to the synthesis of protein isoforms of 1207 and 1130 amino acids, respectively. The three CIITA isoforms have apparent molecular weights of 132 kD, 124 kD and 121 kD (Fig.8). All three protein variants exist in vivo. The first CIITA cDNA clone to be isolated corresponded to a CIITA type III mRNA. Therefore, nucleotide and amino acid numbering are generally given with respect to the transcription initiation site of type III mRNA and the translational initiation codon of the 1130 kD isoform.

pI is active in dendritic cells and in IFN-g activated monocytes and macrophages. pII is only found in humans and its significance is unknown. pIII is used in B cells, in activated human T cells and in some types of dendritic cells. pIV is responsive to IFN-g and is constitutively expressed in cTECs. The three types of mRNAs (types I, III and IV) initiated at pI, pIII and pIV encode three different protein isoforms (121, 124 and 132 kD). These proteins differ only at their N-terminal end. Gray bars represent exons. white bars represent mRNAs and black bars represent proteins encoded by these mRNAs. The boundary between the alternative first exons and the shared downstream exons is indicated by a vertical line. The positions of translation initiation codons are indicated.
The N-terminal extensions encoded by the unique first exons of CIITA type I and type III mRNAs may confer specific properties on the corresponding 132 kD and 124 kD CIITA isoforms. It has been suggested that the 132 kD isoform of CIITA, which is mainly expressed in DCs, has an enhanced capacity to activate MHCII transcription compared to the smaller two isoforms [117] [Luc Otten, unpublished results]. In addition, this isoform seems to have an extremely short half-life [our unpublished observations]. It is likely that these features are due to an intrinsic property of the N-terminus unique to this CIITA isoform. A recent study has proposed that the N-terminal extension of the 132 kD isoform displays homology to caspase recruitment domains (CARD) [117], which are found in components of the apoptosis and NF-kB signaling pathways [118,119]. The significance of this is not clear because CIITA is not known to be involved in either of these pathways.
Cell type specific and modulated expression of the MHC2TA gene is controlled by the differential activities of the three promoters. It is thus the sophisticated transcriptional control of the MHC2TA gene that dictates the cell type specific and inducible expression of MHCII genes.
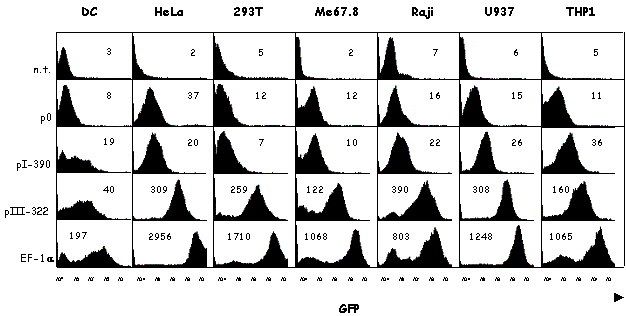
pI and expression of MHC2TA in DC
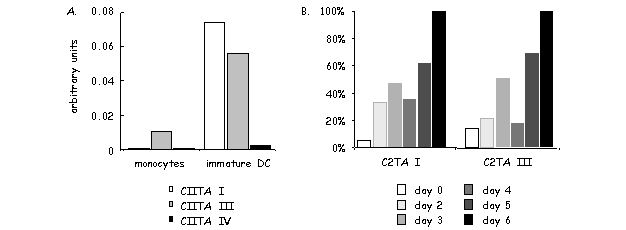
MHC2TA pI is highly specific for DCs. CIITA type I transcripts always represent a preponderant fraction in various DC preparations including ex vivo mouse DCs, mouse bone marrow-derived DCs, long-term mouse DC cultures and human monocyte-derived DCs [116,120-122] [our unpublished data]. However, CIITA type III transcripts are also found in significant amounts in certain DC preparations [121]. Moreover, DC specificity of pI is not as absolute as first thought. pI can for instance also be induced by IFN-g in microglia and peritoneal macrophages [120,122].
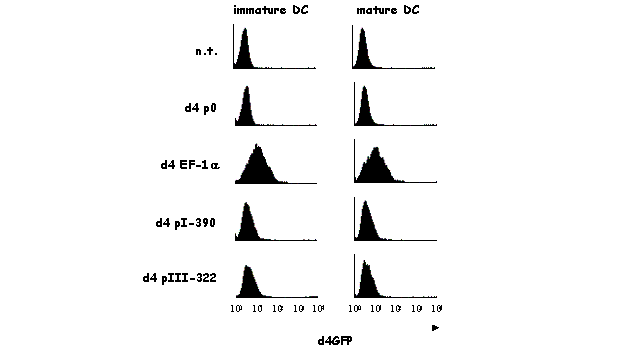
A recent report from our laboratory has characterized in detail the activity of pI (and pIII) during the course of maturation of human monocyte-derived DCs [121]. In response to a variety of stimuli, such as infection by bacteria or viruses, immature DCs are induced to undergo profound changes in their morphology and function. Changes in the expression of MHCII represent a key aspect of this maturation process. The number of MHCII molecules expressed at the cell surface is increased as a result of changes in the intracellular localization and stability of preexisting MHCII protein. In contrast, de novo synthesis of MHCII molecules is shut down. This reduction in MHCII synthesis during DC maturation is a consequence of rapid silencing of the MHC2TA gene. The arrest in CIITA expression is the result of a transcriptional inactivation of the MHC2TA gene. This is mediated by a global repression mechanism implicating histone deacetylation over a large domain spanning the entire MHC2TA regulatory region, including the two promoters (pI and pIII) that are active in DCs.
pIII and expression of MHC2TA in B cells
MHC2TA pIII is used primarily in B cells [116,123], activated human T cells [98] [our unpublished observations] and to a lesser extent in some DC subsets [116,121]. Aberrant CIITA expression in melanoma cells was also reported to be initiated at pIII [124,125]. A 320 bp promoter proximal regulatory region is sufficient for B cell specific activity [116,123]. This region contains five sequence elements that can be shown to be occupied in vivo in B cells in genomic footprinting experiments [126] (Fig.9B). At least two of these elements are critical for proper activity, namely a TEF-2 like element and a binding site for a novel transcription factor [126].
A 5' flanking sequence situated approximately five kb upstream of the transcription initiation site of pIII of the human MHC2TA gene has been reported to confer IFN-g responsiveness [103]. The functional relevance of this sequence is supported by the presence of a DNaseI hypersensitive site at this position in vivo [127]. The putative regulatory region acts as a STAT-1 dependent and IRF-1 independent enhancer [127]. In contrast to these findings in human cells, pIII does not seem to be inducible by IFN-g in primary rat astrocytes [112] and in mouse macrophages [122]. There may thus be a species specific difference in the IFN-g responsiveness of pIII.
Expression of the MHC2TA gene is actively silenced during terminal differentiation of B cells into plasmocytes [90,99]. The sequence elements of pIII that are occupied in normal B cells are completely bare in plasmocytes [90,126]. The human positive regulatory domain I binding factor-1 (PRDI-BF1) [128] and its mouse homologue B lymphocyte-induced maturation protein-1 (Blimp-1) [129] have been proposed to play a crucial role in the repression of pIII in plasmocytes [130,131]. PRDI-BF1/Blimp-1 is upregulated when B cells differentiate into plasmocytes [132]. It has also been shown to drive, at least partially, the final differentiation of B cells into plasmocytes if expressed ectopically in BCL1 lymphoma cells [132]. PRDI-BF1/Blimp-1 can bind specifically to a sequence in the promoter proximal region of pIII [130,131]. However, no occupation of this site is seen in in vivo footprint experiments performed with plasmocytes [126]. The mechanism, by which PRDI-BF1/Blimp-1 silences the MHC2TA gene, is thus not well understood.

a) The regulation of pI is not well understood. The depicted elements represent only potential consensus binding sites. The arrowheads represent protections and enhancements observed in in vivo footprint experiments at the sense (downwards) and antisense (upwards) strands in human dendritic cells. b) pIII contains five cis-regulatory elements defined by in vivo footprint experiments (arrowheads). They are called site A, site B, ARE-2, ARE-1 and site C. In vitro binding studies have revealed that ARE-1 may be bound by AML2 in B and T cells, ARE-2 by a member of the CREB/ATF protein family in B and T cells, site A by NF-1 in B cells and site C by PRDI-BF1 in plasmocytes. c) The regulation of IFN-g induced pIV expression is well understood. Binding of IFN-g to its receptor induces JAK-1 and JAK-2 activation and thus STAT-1 phosphorylation, dimerization and translocation to the nucleus. STAT-1 binds cooperatively with USF-1 to a composite GAS/E box motif. STAT-1 also activates expression of IRF-1, which then binds to its cognate site in pIV. The arrowheads depict protections and enhancement observed in in vivo footprint experiments. The mechanism mediating expression of pIV in cTECs is not known.
pIV and IFN-g induced MHC2TA expression
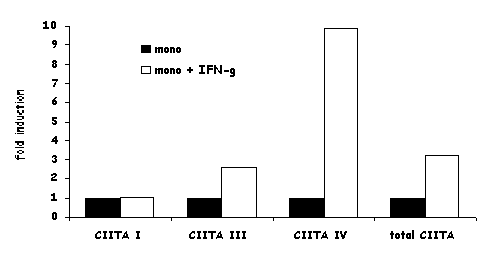
MHC2TA pIV is induced by IFN-g in most cell types, including cells of the monocyte/macrophage lineage, endothelial cells, fibroblasts, astrocytes and cTECs [103,105,110,116,127,133]. A 300 bp promoter proximal region is sufficient for the IFN-g response [116,127,133]. This region contains a GAS element, an E box and an IRF-1 binding site (Fig.9C), all three of which are required for induction in most cell types [103,110,116,133,134]. The former two are bound cooperatively by STAT-1 and upstream regulatory factor-1 (USF-1) [103,110,133]. STAT-1 is activated and translocated to the nucleus by the classical IFN-g signal transduction pathway. USF-1 is a constitutively expressed member of the basic helix-loop-helix/leucine zipper family. In rat astrocytes, STAT-1 is dispensable for IFN-g induced activation of pIV [112]. This may represent a species or cell type specific exception to the general requirement for STAT-1 in IFN-g induced pIV activity. The IRF-1 binding site of pIV is occupied by IRF-1. IRF-1 synthesis itself is induced by IFN-g. Thus, the dependence of CIITA on IRF-1 explains the delayed kinetics of CIITA induction by IFN-g. [89]. One group has proposed that IRF-1 and IRF-2 bind cooperatively to pIV [135], and found a reduction of CIITA expression in IRF-2 knockout mice [136]. IRF-2 may thus also be involved in activation of pIV.
IFN-g activated expression of CIITA can be suppressed by a number of different stimuli. TGF-b markedly attenuates IFN-g induced CIITA expression. The inhibitory mechanism involves suppression at the level of MHC2TA transcription [101,102,104]. Surprisingly, TGF-b does not affect IFN-g induced phosphorylation of JAK-1, JAK-2 and STAT-1. Nor does it interfere with binding of STAT-1, USF-1 or IRF-1 to pIV of the MHC2TA gene [137,138]. Moreover, TGF-b even inhibits basal non-induced expression levels of pIV [103,139]. Finally ,activity of the putative IFN-g response element situated upstream of pIII is also inhibited by TGF-b [7]. A recent report has implicated Smad-3 in the inhibitory effect of TGF-b on CIITA expression [138]. IL-1, IL-4 and IL-10 have also been shown to exert an inhibitory effect similar to TGF-b on CIITA transcription in human astrocytes (IL-1) [105] and in mouse microglia cells (IL-4, IL-10) [104]. The role of IL-4 mediated CIITA inhibition seems to be cell type dependent.
IFN-g induced gene activation is generally a transient event. Suppressor of cytokine signaling-1 (SOCS-1) has been shown to be induced by IFN-g and this protein negatively regulates the IFN-g signal transduction pathway by binding to JAK-2 and inhibiting its kinase activity [140,141]. SOCS-1 can thus also suppress IFN-g activated expression of pIV of the MHC2TA gene [134].
Trophoblasts lack MHCII and CIITA expression in order to evade maternal immune recognition. They do not respond to IFN-g, although MHCII can be induced by ectopic expression of CIITA in these cells [94,100]. Recently, two laboratories have reported that suppression of CIITA expression in trophoblasts is due to methylation of pIV [142,143].
Lessons from pIV knockout mice
Mhc2ta pIV knockout mice have been generated in our laboratory [122]. These mice allowed us to define precisely the cell type specificity of pIV in vivo. pIV -/- mice exhibit selective abrogation of IFN-g induced MHCII expression on a wide variety of non-hematopoietic cells, including endothelia, epithelia, astrocytes and fibroblasts. The cTECs of these mice are also MHCII negative. In contrast, constitutive and inducible MHCII expression is unaffected on professional APCs including B cells, DCs and IFN-g activated cells of the monocyte/macrophage lineage.
The phenotype of the pIV -/- mice demonstrates that professional APCs do no depend on pIV for IFN-g inducible expression of CIITA. Ex vivo macrophages from control mice use both, pI and pIV in response to IFN-g. Macrophages from pIV deficient animals express similar amounts of total CIITA mRNA in response to IFN-g but transcripts are derived exclusively from pI. It remains unknown, how IFN-g activates pI. No IFN-g responsive sequences have been identified in the vicinity of pI. It is thus possible that IFN-g may affect pI indirectly as a consequence of macrophage activation. The answer to this question awaits further dissection of the regulatory mechanism controlling pI.
The finding that constitutive MHCII expression on cTECs is abolished was completely unexpected. The loss of MHCII molecules on cTECs in the pIV knockout mice results in the abrogation of positive selection of CD4+ T cells in the thymus. CD4+ T cell counts in these mice are strongly reduced in the thymus and in the periphery. pIV is thus essential for positive selection of CD4+ T cells.
Isolated cTECs loose MHCII expression if they are not maintained in three-dimensional re-aggregate thymus organ cultures [144,145]. This indicates that a stimulus must be required to maintain constitutive MHCII expression on cTECs. This stimulus could be provided either by cell-cell interactions between the cTECs or by a short acting soluble factor produced by the cTECs. The precise nature of the signal required for MHCII expression in cTECs as well as its mode of action is still unknown. The phenotype of the pIV-/- mice indicates that this signal functions by activating pIV of the MHC2TA gene. However, it must achieve this via a mechanism that is independent of the IFN-g signaling pathway, because knockout mice lacking key components of this pathway, such as the IFN-g receptor, STAT-1 and IRF-1, have normal MHCII expression on cTEC and normal positive selection of CD4+ T cells.
The Mhc2ta pIV knockout mice have provided definitive evidence that differential CIITA promoter usage does indeed play an important physiological role. They have also allowed us to draw a more accurate picture of the differential promoter usage. Cells of myeloid origin (DCs, monocytes/macrophages) use mainly pI for constitutive or IFN-g induced CIITA expression. Constitutive CIITA expression in cells of lymphoid origin (B cells, T cells) is driven by pIII. Finally, pIV is indispensable for IFN-g activated expression in non-hematopoietic cells and for expression in cTECs.
Structure and function of CIITA
Structure of CIITA
Given the key role of CIITA as the master regulator of MHCII expression, a considerable amount of effort has been devoted to the elucidation of its structure and mode of action. Analysis of the primary sequence of CIITA has revealed four major features of interest, namely an N-terminal acidic domain, an adjacent proline/serine/threonine rich (PST) region, a central GTP binding domain and a series of leucine rich repeats (LRR) at the C-terminus [9,64] (Fig.7A). Several other proteins have been found to share a similar domain structure. In particular, the nucleotide binding domain and the leucine rich repeats are conserved in a number of proteins [64]. Intriguingly, these proteins have functions very distinct from that of CIITA. They include for instance Nod-1, which induces activation of caspase-9 and NF-kB, and certain plant resistance proteins [119,146]. The similarity in the domain structure between CIITA and these unrelated proteins may indicate a yet unknown analogy in their mode of action.
Recruitment of CIITA to the MHCII enhanceosome
In vivo association of CIITA with MHCII and related promoters was recently demonstrated by chromatin immunoprecipitation experiments [63,147]. Moreover, DNA dependent co-immunoprecipitation assays and pull-down experiments using immobilized promoter templates have demonstrated a direct physical interaction between CIITA and the MHCII enhanceosome [63]. This interaction requires the integrity of the enhanceosome and depends on multiple, synergistic protein-protein interactions with the multiple factors constituting the enhanceosome.
CIITA has been demonstrated to interact with several components of the MHCII enhanceosome. An interaction with RFX5 was first revealed in a yeast-two-hybrid system [148]. In vitro interactions were subsequently also demonstrated with RFX5, RFXANK, NF-YB, NF-YC and CREB [69,84,149]. The regions of CIITA that are required for recruiting CIITA to the enhanceosome [63,149] and for co-immunoprecipitation with these proteins [84] have been partially mapped.
Activation of transcription by CIITA
CIITA activates transcription of MHCII genes when recruited to the enhanceosome. The regions of the protein that are essential for this transactivation have been mapped to the N-terminal acidic and PST domains [150-152]. These domains are believed to constitute transcription activation domains and they resemble the activation domains found in other transcription factors. When fused to a GAL4 DNA binding domain, the N-terminus of CIITA can activate transcription of a reporter gene [150]. Moreover, the transcription activation domains of VP16 and HSV1a transducing factor can substitute for the N-terminus of CIITA [150,151]. This substitution is only partial, indicating that the N-terminus of CIITA may also support additional functions.
Deletion of the acidic and (or) PST domains results in dominant negative mutants that repress the activity of wild type CIITA in B cells and fibroblasts [152-156]. In vitro promoter pull down experiments have shown that these dominant negative mutants of CIITA are recruited more efficiently to MHCII promoters than wild type CIITA [63]. This suggests that their dominant negative phenotype is due at least in part to an increased affinity for the enhanceosome.
Various general transcription factors or coactivators can interact with the N-terminus of CIITA (Fig.10). The TAFII32 and TAFII70 subunits of the TFIID complex, TFIIB, TFIIH, pTEFb, CBP and pCAF have been shown to be able to interact with this region [154,157-160]. This suggests that the N-terminus of CIITA acts as a transcriptional activation domain recruiting factors involved in transcription initiation (TFIID, TFIIB), promoter clearance and transcription elongation (TFIIH, pTEFb), or chromatin remodeling (CBP, pCAF). The interactions of CIITA with CBP and pCAF have been addressed in greater detail. First, CBP and pCAF can both increase, synergistically with CIITA, the expression of a HLA-DRA reporter gene [158,161,162]. Surprisingly, this cooperativity has been shown to be independent of the histone acetyltransferase (HAT) activity of CBP and pCAF [162]. Second, the synergy between CIITA and CBP is inhibited by a dominant negative variant of CBP [158]. Finally, when overexpressed, CIITA can sequester CBP and thus downregulate other CBP dependent genes [158] (see below) [163].

Interactions with several enhanceosome components tether CIITA to MHCII and related promoters. Once tethered to the promoter, CIITA is believed to activate transcription by recruiting other factors including TFIIB, TFIID, TFIIH, pTEFb, CBP and pCAF. These factors are involved in transcription initiation (TFIIB, TFIID), promoter clearance and transcription elongation (TFIIH, pTEFb) and chromatin remodeling (CBP, pCAF). Protein-protein interactions are indicated by double headed arrows.
Acetylation of lysines in the N-terminal tails of histones H3 and H4 is generally associated with increased transcriptional activity of eukaryotic genes [164-166]. In MHCII and related genes, increased acetylation of histones H3 and H4 correlates with binding of CIITA to the enhanceosome in various cell types [85,147]. The high acetylation status observed in the wild type B cell line Raji is strongly reduced in the CIITA deficient cell line RJ2.2.5 [85,147]. In IFN-g induced HeLa cells, the kinetics of the increase in H3 and H4 acetylation correlates well with the binding of CIITA to the HLA-DRA promoter [147]. Both processes are very dynamic. Binding of CIITA and acetylation start to increase within a few hours after induction by IFN-g, are maximal after 24 hours and then decrease. Acetylation of the HLA-DRA promoter is pronounced at the promoter proximal region [85,147] but is also seen as far as 15 kb upstream of the transcription initiation site [K.Masternak, unpublished results] Multiple HAT activities may be involved in histone acetylation at MHCII promoters. These include the CBP and pCAF co-activators that can bind to CIITA. It has also been proposed that CIITA itself contains an intrinsic acetyl transferase activity [167].
The GTP binding domain and the leucine rich repeat region
CIITA is the first transcription factor known to have a GTP binding domain. This GTP binding domain (amino acids 421 to 561) is composed of a Walker A motif (also called P loop) involved in phosphate binding, a magnesium binding site and a guanine coordination site. This tripartite motif is essential for GTP binding and CIITA function [152,168]. Mutants of the GTP binding domain fail to translocate to the nucleus (see below). Mice bearing a deletion spanning the GTP binding domain of the Mhc2ta gene display a phenotype similar to that of other CIITA knockout mice [45].
LRR domains are known to mediate protein-protein interactions and are found in many different classes of proteins. The LRRs found at the C terminus of CIITA (amino acids 988 to 1097) are essential for its function [149,169]. Partial deletion of these LRRs generates a CIITA mutant displaying a dominant negative phenotype [169].
Dimerization of CIITA
Several laboratories have reported that CIITA can bind to itself via intra- or intermolecular contacts [170-172]. However, there is no uniform consensus on the precise regions involved in this self-association. The central region encompassing the GTP binding site may contact the acidic and PST region [170,172] or may instead bind to the C-terminus [171,172]. LXXLL motifs residing in the GTP binding domain may play a crucial role for self-association [172]. In vitro translated CIITA does not dimerize, indicating that additional factors or modifications may be involved [170-172]. Finally, the precise role of self-association of CIITA is not certain. It may play a role in subcellular localization of CIITA [170]. It should be mentioned here that many of the results described above have been obtained under conditions of overexpression of CIITA and should therefore be interpreted with caution.
Nuclear localization of CIITA
Depending on the cell type and the method of detection, CIITA is either found in both the nucleus and cytoplasm or found predominantly in the nucleus [149,160,168,170,173,174]. As mentioned above for dimerization, nuclear localization studies have been done mainly with overexpressed CIITA. Three potential NLS's have been identified in CIITA (Fig.7A). NLS1 (amino acids 141 to 159) resembles a bipartite NLS [160]. Nuclear localization mediated by NLS1 is affected positively by CBP and pCAF, but this is independent of their HAT activity [160,162]. NLS2 has been localized between amino acids 405 and 414 [174]. It displays similarity to the classical SV40 NLS. Deletion of NLS2 results in cytoplasmic localization and the loss of transactivation by CIITA [174]. Finally, NLS3 was identified thanks to a BLS patient expressing a CIITA mutant that lacks an exon spanning amino acids 940 to 963 [173]. This exon contains a RFLKK motif that functions as a NLS. The loss of this exon abolishes nuclear import of CIITA [173]. In addition to the three NLS's, GTP binding and integrity of the LRRs have been found to be crucial for proper nuclear import of CIITA [149,168,175]. Finally, nuclear export sequences (NES's) have also been found to influence the subcellular distribution of CIITA. Two regions in CIITA have been found to be crucial for nuclear export and can bind to the export receptor CRM1 [170]. The presence of both NLS's and NES's suggests that CIITA shuttles back and forth between the nuclear and cytoplasmic compartments.
The role of CIITA and the enhanceosome in MHCI expression
In addition to their crucial role in MHCII gene expression, both CIITA and the MHCII enhanceosome contribute to the regulation of MHCI genes [63,176-178]. The W, X, X2 and Y sequence elements characteristic of MHCII promoters are also present in the promoter proximal regions of the MHCI and b2m genes. The X2 element in MHCI promoters corresponds to a sequence that was first called site a. The latter was defined as a sequence that has homology to the cAMP response element (CRE) and is bound by members of the ATF/CREB family. The Y box of MHCI promoters was initially called enhancer B. CIITA and the RFX complex contribute to the expression of MHCI genes by interacting with the W-X-X2-Y region of MHCI promoters. This has been demonstrated by chromatin immunoprecipitation experiments and by transactivation assays [63,176-178]. The contribution of CIITA and RFX to the activation of MHCI promoters explains the finding that BLS patients frequently have reduced levels of MHCI expression [11,12].
Compared to their essential role at MHCII promoters, CIITA and RFX contribute only relatively weakly to the expression of MHCI genes. This is evident from the phenotype of BLS patients, which exhibit a complete absence of MHCII expression but retain strong, albeit reduced, levels of MHCI expression [11,12]. The same is also true for mouse models of the human disease [42-45]. CIITA deficient mice do not display significantly reduced MHCI expression. In RFX5 deficient mice MHCI expression is reduced two to ten fold in certain cell types (B and T lymphocytes) [our unpublished data]. The fact that RFX and CIITA are less important for MHCI expression is readily explained by the presence of additional key regulatory sequences present in the promoters of MHCI and b2m genes.
Additional targets of CIITA
Although MHCII and related genes are undoubtedly the major target genes of CIITA, it has recently also been implicated in other systems. For several genes it has been proposed that CIITA functions as a negative regulator. These include the IL-4 gene, the Fas ligand gene and the collagen a2 (I) gene [163,179,180]. In all three examples, sequestration of CBP by CIITA has been proposed to account for the repression. The same mechanism may also be implicated in downregulation of the thymidine kinase and cyclin D1 genes [163].
IL-4, a Th2-type cytokine, was found to be aberrantly expressed in Th1 cells derived from CIITA knock-out mice [179]. Moreover, ectopic expression of CIITA in Th2 cells from CIITA knockout mice was found to suppress the expression of Th2 cytokines [179]. Taken together, these results suggested that CIITA can inhibit expression of the IL-4 gene. The negative effect of CIITA was proposed to be due to a competition between NF-AT, an essential IL-4 gene transcription factor, and CIITA for binding to CBP [181]. Whether repression of IL-4 expression by CIITA actually occurs in vivo remains to be demonstrated. In this respect it should be mentioned that ectopic expression of CIITA at physiological levels in transgenic mice does not affect IL-4 expression in Th2 cells [Luc Otten, unpublished data].
NF-AT is also an important transcription factor for Fas ligand expression. As for the IL-4 gene, the activity of NF-AT on the FasL promoter was shown to be repressed by overexpression of CIITA [180].
The collagen a2(I) gene was recently recognized as a new target for repression by CIITA [163]. The mechanism proposed for downregulation of this gene again involves binding of CIITA to CBP.
The promoter of the HIV provirus recently attracted attention as a novel target of transcriptional regulation by CIITA. Saiffudin et al. reported that expression of CIITA increases LTR promoter activity and HIV replication in fibroblast and T cell lines [182,183]. This was speculated to be relevant because CIITA is expressed in activated human T cells and macrophages, both of which are primary targets of HIV infection. However, in other studies HIV infection and LTR activity were found to be reduced in CIITA positive B and T cell lines [184,185]. It is therefore presently not clear exactly how CIITA affects the level of transcription from the HIV promoter under physiological conditions.
The viral transactivator Tat was reported to be able to downregulate MHCII expression in HIV infected or Tat transfected fibroblasts and T cell lines [159,184,186]. MHCII protein and mRNA levels were reduced in these cells while CIITA expression levels were not affected. Conversely, overexpression of CIITA was found to inhibit Tat-mediated transactivation of the HIV-1 LTR [184]. Tat and CIITA can both bind to the same surface in cyclin T1, one of the subunits of the elongation factor pTEFb. It was therefore proposed that CIITA and Tat compete for binding to pTEFb [159,184]. In addition, a mechanism independent of pTEFb has also been implicated in inhibition of CIITA activity by Tat [187]. Taken together, these findings suggest that the actions of Tat and CIITA may be mutually antagonistic. It is likely that the precise outcome of the reciprocal actions of Tat and CIITA will depend on the relative concentrations of the two proteins and possibly on the cell type in which they are expressed. It remains to be determined if Tat is inhibited by or inhibits CIITA in vivo during the course of infection by HIV.
CIITA as a target of pathogens
Pathogens have developed a wide variety of strategies to escape immune surveillance by their hosts. In order to inhibit the establishment of a protective immune response, several bacteria and viruses have acquired the ability to downregulate MHCII expression and thus prevent activation of specific CD4+ T cells. They achieve this by interfering with the function or expression of CIITA. The intracellular bacterium Chlamydia downregulates CIITA expression by inducing degradation of USF-1, an ubiquitous factor that is required for the activation of pIV of the MHC2TA gene [108]. Infection with Mycobacterium bovis bacillus Calmette-Guérin also downregulates CIITA expression, but the precise mechanisms that is involved remains unknown [107]. Varizella zoster virus inhibits IFN- g induced transcription of the MHC2TA gene by interfering with the IFN-g signaling pathway. It blocks STAT1a and JAK2 protein expression and therefore transcription of the downstream IRF-1 and CIITA genes [109]. Human Cytomegalovirus has also developed mechanisms to modulate IFN-g induced CIITA expression. Infection with this virus can direct JAK-1 protein to the proteasome for degradation or affect the IFN-g signaling pathway downstream of STAT phosphorylation and nuclear translocation [106,188]. Finally, as mentioned above, the HIV Tat protein can inhibit MHCII expression by interfering with the function of CIITA [159,184,186]. Taken together, these findings demonstrate that many pathogens have acquired a means to target CIITA in order to escape immune recognition and establish persistence. It will be of great interest to further our understanding of the strategies used by pathogens to interfere with CIITA and MHCII expression. This will contribute to the design of new approaches for fighting these pathogens.
Acknowledgments
The authors would like to thank all past and current members of the laboratory for their contributions to the work reviewed here. The work performed in the laboratory of the authors was supported by the Swiss National Science Foundation, the NCCR-NEURO, the Gabriella Giorgi-Cavaglieri Foundation, the Ernst and Lucie Schmidheiny Foundation, the Roch Research Foundation and the Novartis Stiftung.
References
- tructrucCresswell P: Assembly, transport, and function of MHC class II molecules. Annu.Rev.Immunol. 1994, 12: 259-293.
- tructrucViret C and Janeway CAJ: MHC and T cell development. Reviews in Immunogenetics 1999, 1: 91-104.
- tructrucBenoist C and Mathis D: Regulation of major histocompatibility complex class II genes: X, Y and other letters of the alphabet. Ann.Rev.Immunol. 1990, 8: 681-715.
- tructrucGlimcher LH and Kara CJ: Sequences and factors: A guide to MHC class-II transcription. Annu.Rev.Immunol. 1992, 10: 13-49.
- tructrucTing JP and Baldwin AS: Regulation of MHC gene expression. Curr.Opin.Immunol. 1993, 5: 8-16.
- tructrucBoss JM: Regulation of transcription of MHC class II genes. Curr.Opin.Immunol. 1997, 9: 107-113.
- tructrucMasternak K, Muhlethaler-Mottet A, Villard J, Peretti M, Reith W: Molecular genetics of the Bare lymphocyte syndrome. Rev.Immunogenet. 2000, 2: 267-282.
- tructrucWaldburger JM, Masternak K, Muhlethaler-Mottet A, Villard J, Peretti M, Landmann S, Reith W: Lessons from the bare lymphocyte syndrome: molecular mechanisms regulating MHC class II expression. Immunol.Rev. 2000, 178: 148-165.
- tructrucReith W and Mach B: The bare lymphocyte syndrome and the regulation of mhc expression. Annu.Rev.Immunol. 2001, 19: 331-373.
- tructrucGuardiola J and Maffei A: Control of MHC class II gene expression in autoimmune, infectious, and neoplastic diseases. Crit.Rev Immunol. 1993, 13: 247-268.
- tructrucKlein C, Lisowska-Grospierre B, LeDeist F, Fischer A, Griscelli C: Major histocompatibility complex class II deficiency: clinical manifestations, immunologic features, and outcome. J.Pediatr. 1993, 123: 921-928.
- tructrucGriscelli C, Lisowska-Grospierre B, Mach B: Combined immunodeficiency with defective expression in MHC class II genes. In Immunodeficiencies. F.S.Rosen and M.Seligman, editors. Harwood Academic Publishers, Chur, Switzerland, 1993, 141-154.
- tructrucElhasid R and Etzioni A: Major histocompatibility complex class II deficiency: a clinical review. Blood Rev. 1996, 10: 242-248.
- tructrucMach B, Steimle V, Martinez-Soria E, Reith W: Regulation of MHC class II genes: Lessons from a disease. Annu.Rev.Immunol. 1996, 14: 301-331.
- tructrucTouraine JL, Marseglia GL, Betuel H, Souillet G, Gebuhrer L: The bare lymphocyte syndrome. Bone Marrow Transplant. 1992, 9 Suppl 1: 54-56.
- tructrucPrimary immunodeficiency diseases: report of a WHO scientific group. Immunodefic.Rev. 1992, 3: 195-236.
- tructrucLisowska-Grospierre B , Fondaneche MC, Rols MP, Griscelli C, Fischer A: Two complementation groups account for most cases of inherited MHC class II deficiency. Hum.Mol.Genet. 1994, 3: 953-958.
- tructrucTouraine J L, Betuel H, Souillet G: Combined immunodeficiency disease associated with absence of cell surface HLA A and B antigen. J.Pediatr. 1978, 93: 47-51.
- tructrucSchuurmann RKB, Van Rood JJ, Vossen JM, Schellekens PTA, Feltkamp-Vroom TM, Doyer E, Gmelig-Meyling F, Visser HKA: Failure of Lymphocyte-Membrane HLA A and B Expression in Two Siblings With Combined Immunodeficiency. Clin.Immunol.Immunopathol. 1979, 14: 418-434.
- tructrucGriscelli C, Durandy A, Virelizier JL, Hors J, Lepage V, Colombani J: Impaired cell-to-cell interaction in partial combined immunodeficiency with variable expression of HLA antigens. In Primary Immunodeficiencies. M.Seligman and W.H.Hitzig, editors. Elsevier/North Holland, Amsterdam, 1980, 499-503.
- tructrucKuis W, Roord J, Zegers BJM, Schuurmann RKB, Heijnen CJ, Baldwin WM, Goulmy E, Claas F, van de Griend RJ, Rijkers GT, Van Rood JJ, Vossen JM, Ballieux RE, Stoop RJ: Clinical and Immunological Studies in a Patient With the 'Bare Lymphocyte' Syndrome. In Bone Marrow Transplantation in Europe. J.L.Touraine, E.Gluckman, and C.Griscelli, editors. Amsterdam:Excerpta Medica, 1981, 201-208.
- tructrucLisowska-Grospierre B, Durandy A, Virelizier JL, Fischer A, Griscelli C: Combined Immunodeficiency with Defective Expression of HLA: Modulation of an Abnormal HLA Synthesis and Functional Studies. Birth Defects 1983, 19: 87-92.
- tructrucHadam MR, Dopfer R, Dammer G, Peter HH, Schlesier M, Müller C, Niethammer D: Defective expression of HLA-D-region determinants in children with congenital agammaglobulinemia and malabsorption: a new syndrome. In Histocompatibility Testing. E.D.Albert, M.P.Baur, and W.R.Mayr, editors. Springer-Verlag, Berlin Heidelberg, 1984, 645-650.
- tructrucReith W, Steimle V, Lisowska-Grospierre B, Fischer A, Mach B: Molecular Basis of Major Histocompatibility Class II Deficiency. In Primary Immunodeficiency Diseases, a Molecular and Genetic Approach. H.Ochs, J.Puck, and E.Smith, editors. Oxford University Press, New York, 1999, 167-180.
- tructrucNocera A, Barocci S, Depalma R, Gorski J: Analysis of Transcripts of Genes Located Within the HLA-D Region in B-Cells from an HLA-Severe Combined Immunodeficiency Individual. Hum.Immunol 1993, 38: 231-234.
- tructrucKern I, Steimle V, Siegrist CA, and Mach B: The two novel MHC class II transactivators RFX5 and CIITA both control expression of HLA-DM genes. Int.Immunol 1995, 7: 1295-1299.
- tructrucRieux Laucat F, Le Deist F, Selz F, Fischer A, de Villartay JP: Normal T cell receptor V beta usage in a primary immunodeficiency associated with HLA class II deficiency. Eur.J.Immunol. 1993, 23: 928-934.
- tructrucReith W, Satola S, Herrero Sanchez C, Amaldi I, Lisowska-Grospierre B, Griscelli C, Hadam MR, Mach B: Congenital immunodeficiency with a regulatory defect in MHC class II gene expression lacks a specific HLA-DR promoter binding protein, RF-X. Cell 1988, 53: 897-906.
- tructrucde Preval C, Lisowska-Grospierre B, Loche M, Griscelli C, Mach B: A trans-acting class II regulatory gene unlinked to the MHC controls expression of HLA class II genes. Nature 1985, 318: 291-293.
- tructrucHume CR and Lee JS: Congenital immunodeficiencies associated with absence of HLA class II antigens on lymphocytes result from distinct mutations in trans-acting factors. Hum.Immunol. 1989, 26: 288-309.
- tructrucBenichou B and Strominger JL: Class II-antigen-negative patient and mutant B-cell lines represent at least three, and probably four, distinct genetic defects defined by complementation analysis. Proc.Natl.Acad.Sci.USA 1991, 88: 4285-4288.
- tructrucSeidl C, Saraiya C, Osterweil Z, Fu YP, Lee JS: Genetic Complexity of Regulatory Mutants Defective for HLA Class- II Gene-Expression. J.Immunol. 1992, 148: 1576-1584.
- tructrucHasegawa SL, Riley JL, Sloan III JH, Boss JM: Protease treatment of nuclear extracts distinguishes between class II MHC X1 box DNA-binding proteins in wild-type and class II-deficient B cells. J.Immunol. 1993, 150: 1781-1793.
- tructrucRiley JL and Boss JM: Class II MHC transcriptional mutants are defective in higher order complex formation. J.Immunol. 1993, 151: 6942-6953.
- tructrucSteimle V, Otten LA, Zufferey M, Mach B: Complementation cloning of an MHC class II transactivator mutated in hereditary MHC class II deficiency. Cell 1993, 75: 135-146.
- tructrucSteimle V, Durand B, Barras E, Zufferey M, Hadam MR, Mach B, Reith W: A novel DNA binding regulatory factor is mutated in primary MHC class II deficiency (Bare Lymphocyte Syndrome). Genes & Dev. 1995, 9: 1021-1032.
- tructrucDurand B, Sperisen P, Emery P, Barras E, Zufferey M, Mach B, Reith W: RFXAP, a novel subunit of the RFX DNA binding complex is mutated in MHC class II deficiency. EMBO J 1997, 16: 1045-1055.
- tructrucMasternak K, Barras E, Zufferey M, Conrad B, Corthals G, Aebersold R, Sanchez JC, Hochstrasser DF, Mach B, Reith W: A gene encoding a novel RFX-associated transactivator is mutated in the majority of MHC class II deficiency patients. Nat.Genet 1998, 20: 273-277.
- tructrucNagarajan UM, Louis-Plence P, DeSandro A, Nilsen R, Bushey A, Boss JM: RFX-B is the gene responsible for the most common cause of the bare lymphocyte syndrome, an MHC class II immunodeficiency. Immunity 1999, 10: 153-162.
-
*tructrucWiszniewski W, Fondaneche MC, Le Deist F, Kanariou M, Selz F, Brousse N, Steimle V, Barbieri G, Alcaide-Loridan C, Charron D, Fischer A, Lisowska-Grospierre B: Mutation in the class II trans-activator leading to a mild immunodeficiency. J.Immunol. 2001, 167: 1787-1794.
The group A BLS patients described in this paper display an attenuated clinical phenotype or even an asymptomatic clinical course despite their profound defect in the expression of MHCII genes. More and more patients with such an attenuated phenotype are being identified indicating that the BLS disease may be more frequent than previously thought. - tructrucQuan V, Towey M, Sacks S, Kelly AP: Absence of MHC class II gene expression in a patient with a single amino acid substitution in the class II transactivator protein CIITA. Immunogenetics 1999, 49: 957-963.
- tructrucChang CH, Guerder S, Hong SC, van Ewijk W, Flavell R: Mice lacking the MHC class II transactivator (CIITA) show tissue- specific impairment of MHC class II expression. Immunity 1996, 4: 167-178.
- tructrucClausen BE, Waldburger JM, Schwenk F, Barras E, Mach B, Rajewsky K, Forster I, Reith W: Residual MHC class II expression on mature dendritic cells and activated B cells in RFX5-deficient mice. Immunity 1998, 8: 143-155.
- tructrucWilliams GS, Malin M, Vremec D, Chang CH, Boyd R, Benoist C, Mathis D: Mice lacking the transcription factor CIITA--a second look. Int Immunol. 1998; 10: 1957-1967.
- tructrucItoh-Lindstrom Y, Piskurich JF, Felix NJ, Wang Y, Brickey WJ, Platt JL, Koller BH, Ting JP: Reduced IL-4-, lipopolysaccharide-, and IFN-gamma-induced MHC class II expression in mice lacking class II transactivator due to targeted deletion of the GTP-binding domain. J.Immunol. 1999, 163: 2425-2431.
- tructrucSchuurman HJ, van de Wijngaert FP, Huber JP, Schuurman RK, Zegers BJ, Roord JJ, Kater L: The thymus in 'bare lymphocyte' syndrome: significance of expression of major histocompatibility complex antigens on thymic epithelial cells in intrathymic T-cell maturation. Hum.Immunol. 1985, 13: 69-82.
- tructrucCardell S, Tangri S, Chan S, Kronenberg M, Benoist C, Mathis D:: CD1-restricted CD4+ T cells in major histocompatibility complex class II-deficient mice. J.Exp.Med. 1995, 182: 993-1004.
- tructrucvan Eggermond MC, Rijkers GT, Kuis W, Zegers BJ, van den Elsen PJ: T cell development in a major histocompatibility complex class II- deficient patient. Eur.J.Immunol. 1993, 23: 2585-2591.
- tructrucHenwood J, van Eggermond MCJA, van Boxel-Dezaire AHH, Schipper R, den Hoedt M, Peijnenburg A, Sanal Ö, Ersoy F, Rijkers GT, Zegers BJM, Vossen JM, van Tol MJD, van den Elsen PJ: Human T Cell Repertoire Generation in the Absence of MHC Class II Expression Results in a Circulating CD4+CD8- Population with Altered Physicochemical Properties of Complementarity-Determining Region 3. J.Immunol. 1996, 156: 895-906.
- tructrucDurandy A, Cerf-Bensussan N, Dumez Y, Griscelli C: Prenatal Diagnosis of Severe Combined Immunodeficiency With Defective Synthesis of HLA Molecules. Prenatal Diagnosis 1987, 7: 27-34.
- tructrucKlein C, Cavazzana-Calvo M, Le Deist F, Jabado N, Benkerrou M, Blanche S, Lisowska-Grospierre B, Griscelli C: Bone marrow transplantation in major histocompatibility complex class II deficiency: a single-center study of 19 patients. Blood 1995, 85: 580-587.
- tructrucBonduel M, Pozo A, Zelazko M, Raslawski E, Delfino S, Rossi J, Figueroa C, Sackmann MF: Successful related umbilical cord blood transplantation for graft failure following T cell-depleted non-identical bone marrow transplantation in a child with major histocompatibility complex class II deficiency. Bone Marrow Transplant. 1999, 24: 437-440.
- tructrucCanioni D, Patey N, Cuenod B, Benkerrou M, Brousse N: Major histocompatibility complex class II deficiency needs an early diagnosis: report of a case. Pediatr.Pathol.Lab.Med. 1997, 17: 645-651.
- tructrucHaynes BF, Markert ML, Sempowski GD, Patel DD, Hale LP: The role of the thymus in immune reconstitution in aging, bone marrow transplantation, and HIV-1 infection. Annu.Rev.Immunol. 2000, 18: 529-560.
- tructrucZinkernagel RM and Althage A: On the role of thymic epithelium vs. bone marrow-derived cells in repertoire selection of T cells. Proc.Natl.Acad.Sci.USA 1999, 96: 8092-8097.
- tructrucOstrand-Rosenberg S, Clements VK, Thakur A, Cole GA: Transfection of major histocompatibility complex class I and class II genes causes tumour rejection. J.Immunogenet. 1989, 16: 343-349.
- tructrucBaskar S, Clements VK, Glimcher LH, Nabavi N, Ostrand-Rosenberg S: Rejection of MHC class II-transfected tumor cells requires induction of tumor-encoded B7-1 and/or B7-2 costimulatory molecules. J.Immunol. 1996, 156: 3821-3827.
- tructrucPanelli MC, Wang E, Shen S, Schluter SF, Bernstein RM, Hersh EM, Stopeck A, Gangavalli R, Barber J, Jolly D, Akporiaye ET: Interferon gamma (IFNgamma) gene transfer of an EMT6 tumor that is poorly responsive to IFNgamma stimulation: increase in tumor immunogenicity is accompanied by induction of a mouse class II transactivator and class II MHC. Cancer Immunol.Immunother. 1996, 42: 99-107.
- tructrucMartin BK, Frelinger JG, Ting JP: Combination gene therapy with CD86 and the MHC class II transactivator in the control of lung tumor growth. J.Immunol. 1999, 162: 6663-6670.
- tructrucHasegawa SL and Boss JM: Two B cell factors bind the HLA-DRA X box region and recognize different subsets of HLA class II promoters. Nucleic Acids Res. 1991, 19: 6269-6276.
- tructrucMoreno CS, Emery P, West JE, Durand B, Reith W, Mach B, Boss JM: Purified X2 binding protein (X2BP) cooperatively binds the class II MHC X box region in the presence of purified RFX, the X box factor deficient in the bare lymphocyte syndrome. J.Immunol. 1995, 155: 4313-4321.
- tructrucMantovani R: The molecular biology of the CCAAT-binding factor NF-Y. Gene 1999, 239: 15-27.
- tructrucMasternak K, Muhlethaler-Mottet A, Villard J, Zufferey M, Steimle V, Reith W: CIITA is a transcriptional coactivator that is recruited to MHC class II promoters by multiple synergistic interactions with an enhanceosome complex. Genes Dev. 2000, 14: 1156-1166.
- tructrucHarton JA and Ting JP: Class II transactivator: mastering the art of major histocompatibility complex expression. Mol.Cell Biol. 2000, 20: 6185-6194.
- tructrucEmery P, Durand B, Mach B, Reith W: RFX proteins, a novel family of DNA binding proteins conserved in the eukaryotic kingdom. Nucl.Acids Res. 1996, 24: 803-807.
- tructrucEmery P, Strubin M, Hofmann K, Bucher P, Mach B, Reith W: A consensus motif in the RFX DNA binding domain and binding domain mutants with altered specifity. Mol.Cell.Biol. 1996, 16: 4486-4494.
- tructrucGajiwala KS, Chen H, Cornille F, Roques BP, Reith W, Mach B, Burley SK: Structure of the winged-helix protein hRFX1 reveals a new mode of DNA binding. Nature 2000, 403: 916-921.
- tructrucVillard J, Peretti M, Masternak K, Barras E, Caretti G, Mantovani R, Reith W: A functionally essential domain of RFX5 mediates activation of major histocompatibility complex class II promoters by promoting cooperative binding between RFX and NF-Y. Mol.Cell Biol. 2000, 20: 3364-3376.
- tructrucDeSandro AM, Nagarajan UM, Boss JM: Associations and interactions between bare lymphocyte syndrome factors. Mol.Cell.Biol. 2000, 20: 6587-6599.
- tructrucPeretti M, Villard J, Barras E, Zufferey M, Reith W: Expression of the three human major histocompatibility complex class II isotypes exhibits a differential dependence on the transcription factor RFXAP. Mol.Cell Biol. 2001, 21: 5699-5709.
- tructrucCaretti G, Cocchiarella F, Sidoli C, Villard J, Peretti M, Reith W, Mantovani R: Dissection of functional NF-Y-RFX cooperative interactions on the MHC class II E alpha promoter. J.Biol.Chem. 2000, 302: 539-552.
- tructrucHerrero Sanchez C, Reith W, Silacci P, Mach B: The DNA-binding defect observed in major histocompatibility complex class II regulatory mutants concerns only one member of a family of complexes binding to the X boxes of class II promoters. Mol.Cell Biol. 1992, 12: 4076-4083.
- tructrucMoreno CS, Rogers EM, Brown JA, Boss JM: Regulatory factor X, a bare lymphocyte syndrome transcription factor, is a multimeric phosphoprotein complex. J.Immunol. 1997, 158: 5841-5848.
- tructrucWesterheide SD and Boss JM: Orientation and positional mapping of the subunits of the multicomponent transcription factors RFX and X2BP to the major histocompatibility complex class II transcriptional enhancer. Nucleic Acids Res. 1999, 27: 1635-1641.
- tructrucReith W, Siegrist CA, Durand B, Barras E, Mach B: Function of major histocompatibility complex class II promoters requires cooperative binding between factors RFX and NF-Y. Proc.Natl.Acad.Sci.U.S.A. 1994, 91: 554-558.
- tructrucReith W, Kobr M, Emery P, Durand B, Siegrist CA, Mach B: Cooperative binding between factors RFX and X2bp to the X and X2 boxes of MHC class II promoters. J.Biol.Chem. 1994, 269: 20020-20025.
- tructrucLouis-Plence P, Moreno CS, Boss JM: Formation of a regulatory factor X/X2 box-binding protein/nuclear factor-Y multiprotein complex on the conserved regulatory regions of HLA class II genes. J.Immunol. 1997, 159: 3899-3909.
- tructrucMoreno CS, Beresford GW, Louis-Plence P, Morris AC, Boss JM: CREB regulates MHC class II expression in a CIITA-dependent manner. Immunity 1999, 10: 143-151.
- tructrucGönczy P, Reith W, Barras E, Lisowska-Grospierre B, Griscelli C, Hadam MR, Mach B: Inherited Immunodeficiency with a defect in a Major Histocompatibility Complex class II promoter-binding protein differs in the chromatin structure of the HLA-DRA gene. Mol.Cell.Biol. 1989, 9: 296-302.
- tructrucKara CJ and Glimcher LH: In vivo footprinting of MHC class II genes: bare promoters in the bare lymphocyte syndrome. Science 1991, 252: 709-712.
- tructrucKara CJ and Glimcher LH: Three in vivo promoter phenotypes in MHC class II deficient combined immunodeficiency. Immunogenetics 1993, 37: 227-230.
- tructrucVilen BJ, Cogswell JP, Ting JP: Stereospecific alignment of the X and Y elements is required for major histocompatibility complex class II DRA promoter function. Mol.Cell.Biol. 1991, 11: 2406-2415.
- tructrucVilen BJ, Penta JF, Ting JP: Structural constraints within a trimeric transcriptional regulatory region: Constitutive and Interferon-g inducible expression of the HLA-DRA gene. J.Biol.Chem. 1992, 267: 23728-23734.
- tructrucZhu XS, Linhoff MW, Li G, Chin KC, Maity SN, Ting JP: Transcriptional scaffold: CIITA interacts with NF-Y, RFX, and CREB To cause stereospecific regulation of the class II major histocompatibility complex promoter. Mol.Cell.Biol. 2000, 20: 6051-6061.
-
**tructrucMasternak K and Reith W: Promoter-specific functions of CIITA and the MHC class II enhanceosome in transcriptional activation. EMBO J. 2002, 21: 1379-1388.
This in vivo study demonstrates that the MHCII enhanceosome contributes at some promoters to histone acetylation and transcription factor recruitment in a CIITA-independent manner. - tructrucRigaud G, De Lerma BA, Nicolis M, Cestari T, Ramarli D, Riviera AP, Accolla RS: Induction of CIITA and modification of in vivo HLA-DR promoter occupancy in normal thymic epithelial cells treated with IFN-gamma: similarities and distinctions with respect to HLA-DR-constitutive B cells. J.Immunol. 1996, 156: 4254-4258.
- tructrucWright KL, Chin KC, Linhoff M, Skinner C, Brown JA, Boss JM, Stark GR, Ting JP: CIITA stimulation of transcription factor binding to major histocompatibility complex class II and associated promoters in vivo. Proc.Natl.Acad.Sci.U.S.A 1998, 95: 6267-6272.
- tructrucVillard J, Muhlethaler-Mottet A, Bontron S, Mach B, Reith W: CIITA-induced occupation of MHC class II promoters is independent of the cooperative stabilization of the promoter-bound multi-protein complexes. Int.Immunol. 1999, 11: 461-469.
- tructrucSteimle V, Siegrist CA, Mottet A, Lisowska-Grospierre B, Mach B: Regulation of MHC class II expression by Interferon-gamma mediated by the transactivator gene CIITA. Science 1994, 265: 106-109.
- tructrucSilacci P, Mottet A, Steimle V, Reith W, Mach B: Developmental extinction of major histocompatibility complex class II gene expression in plasmocytes is mediated by silencing of the transactivator gene CIITA. J.Exp.Med. 1994, 180: 1329-1336.
- tructrucChin KC, Mao C, Skinner C, Riley LJ, Wright KL, Moreno CS, Stark GR, Boss JM, Ting JP: Molecular analysis of G1B and G3A IFN-gamma mutants reveals that defects in CIITA or RFX result in defective class II MHC and Ii gene induction. Immunity 1994, 1: 687-697.
- tructrucChang CH and Flavell RA: Class II transactivator regulates the expression of multiple genes involved in antigen presentation. J.Exp.Med. 1995, 181: 765-767.
- tructrucRamassar V, Goes N, Hobart M, Halloran PF: Evidence for the in vivo role of class II transactivator in basal and IFN-gamma induced class II expression in mouse tissue. Transplantation. 1996, 62: 1901-1907.
- tructrucMorris AC, Riley JL, Fleming WH, Boss JM: MHC class II gene silencing in trophoblast cells is caused by inhibition of CIITA expression. Am.J.Reprod.Immunol. 1998, 40: 385-394.
- tructrucOtten LA, Steimle V, Bontron S, Mach B: Quantitative control of MHC class II expression by the transactivator CIITA. Eur.J.Immunol. 1998, 28: 473-478.
- tructrucChang CH, Fontes JD, Peterlin M, Flavell RA: Class II transactivator (CIITA) is sufficient for the inducible expression of major histocompatibility complex class II genes. J.Exp.Med. 1994, 180: 1367-1374.
- tructrucSartoris S, Valle MT, Barbaro AL, Tosi G, Cestari T, D'Agostino A, Megiovanni AM, Manca F, Accolla RS: HLA class II expression in uninducible hepatocarcinoma cells after transfection of AIR-1 gene product CIITA: acquisition of antigen processing and presentation capacity. J.Immunol. 1998, 161: 814-820.
- tructrucHolling TM, van der Stoep N, Quinten E, van den Elsen PJ: Activated Human T Cells Accomplish MHC Class II Expression Through T Cell-Specific Occupation of Class II Transactivator Promoter III. J.Immunol. 2002, 168: 763-770.
- tructrucSartoris S, Tosi G, De Lerma B, Cestari T, Accolla RS: Active suppression of the class II transactivator-encoding AIR-1 locus is responsible for the lack of major histocompatibility complex class II gene expression observed during differentiation from B cells to plasma cells. Eur.J.Immunol. 1996, 26: 2456-2460.
- tructrucMurphy SP and Tomasi TB: Absence of MHC class II antigen expression in trophoblast cells results from a lack of class II transactivator (CIITA) gene expression. Mol.Reprod.Dev. 1998, 51: 1-12.
- tructrucLee YJ, Han Y, Lu HT, Nguyen V, Qin H, Howe PH, Hocevar BA, Boss JM, Ransohoff RM, Benveniste EN: TGF-beta suppresses IFN-gamma induction of class II MHC gene expression by inhibiting class II transactivator messenger RNA expression . J.Immunol. 1997, 158: 2065-2075.
- tructrucNandan D and Reiner NE: TGF-beta attenuates the class II transactivator and reveals an accessory pathway of IFN-gamma action. J.Immunol. 1997, 158: 1095-1101.
- tructrucPiskurich JF, Wang Y, Linhoff, MW, White LC, Ting JP: Identification of distinct regions of 5' flanking DNA that mediate constitutive, IFN-gamma, STAT1, and TGF-beta-regulated expression of the class II transactivator gene. J.Immunol. 1998, 160: 233-240.
- tructrucO'Keefe GM, Nguyen VT, Benveniste EN: Class II transactivator and class II MHC gene expression in microglia: modulation by the cytokines TGF-beta, IL-4, IL-13 and IL-10. Eur.J.Immunol. 1999, 29: 1275-1285.
- tructrucRohn W, Tang LP, Dong Y, Benveniste EN: IL-1 beta inhibits IFN-gamma-induced class II MHC expression by suppressing transcription of the class II transactivator gene. J.Immunol. 1999, 162: 886-896.
- tructrucMiller DM, Rahill BM, Boss JM, Lairmore MD, Durbin JE, Waldman JW, Sedmak DD: Human cytomegalovirus inhibits major histocompatibility complex class II expression by disruption of the Jak/Stat pathway. J.Exp.Med. 1998, 187: 675-683.
- tructrucWojciechowski W, DeSanctis J, Skamene E, Radzioch D: Attenuation of MHC class II expression in macrophages infected with Mycobacterium bovis bacillus Calmette-Guerin involves class II transactivator and depends on the Nramp1 gene. J.Immunol. 1999, 163: 2688-2696.
- tructrucZhong G, Fan T, Liu L: Chlamydia inhibits interferon gamma-inducible major histocompatibility complex class II expression by degradation of upstream stimulatory factor 1. J.Exp.Med. 1999, 189: 1931-1938.
- tructrucAbendroth A, Slobedman B, Lee E, Mellins E, Wallace M, Arvin AM: Modulation of major histocompatibility class II protein expression by varicella-zoster virus. J.Virol. 2000, 74: 1900-1907.
- tructrucDong Y, Rohn WM, Benveniste EN: IFN-gamma regulation of the type IV class II transactivator promoter in astrocytes. J.Immunol. 1999, 162: 4731-4739.
- tructrucHan Y, Zhou ZH, Ransohoff RM: TNF-alpha suppresses IFN-gamma-induced MHC class II expression in HT1080 cells by destabilizing class II trans-activator mRNA. J.Immunol. 1999, 163: 1435-1440.
- tructrucNikcevich KM, Piskurich JF, Hellendall RP, Wang Y, Ting JP: Differential selectivity of CIITA promoter activation by IFN-gamma and IRF-1 in astrocytes and macrophages: CIITA promoter activation is not affected by TNF-alpha. J.Neuroimmunol. 1999, 99: 195-204.
- tructrucLi G, Harton JA, Zhu X, Ting JP: Downregulation of CIITA function by protein kinase a (PKA)-mediated phosphorylation: mechanism of prostaglandin E, cyclic AMP, and PKA inhibition of class II major histocompatibility complex expression in monocytic lines. Mol.Cell Biol. 2001, 21: 4626-4635.
- tructrucDouhan J, Lieberson R, Knoll JHM, Zhou H, Glimcher LH: An isotype-specific activator of major histocompatibility complex (MHC) class II genes that is independent of class II transactivator. J.Exp.Med. 1997, 185: 1885-1895.
- tructrucAccolla RS, Jotterand-Bellomo M, Scarpellino L, Maffei A, Carra G, Guardiola J: aIr-1, a newly found locus on mouse chromosome 16 encoding a trans-acting activator factor for MHC class II gene expression. J.Exp.Med. 1986, 164: 369-374.
- tructrucMuhlethaler-Mottet A, Otten LA, Steimle V, Mach B: Expression of MHC class II molecules in different cellular and functional compartments is controlled by differential usage of multiple promoters of the transactivator CIITA. EMBO J. 1997, 16: 2851-2860.
- tructrucNickerson K, Sisk TJ, Inohara N, Yee CS, Kennell J, Cho MC, Yannie PJ, Nunez G, Chang CH: Dendritic cell-specific MHC class II transactivator contains a caspase recruitment domain that confers potent transactivation activity. J.Biol.Chem. 2001, 276: 19089-19093.
- tructrucHofmann K, Bucher P, Tschopp J: The CARD domain: a new apoptotic signalling motif. Trends Biochem.Sci. 1997, 22: 155-156.
- tructrucInohara N, Koseki T, del Peso L, Hu Y, Yee C, Chen S, Carrio R, Merino J, Liu D, Ni J, Nunez G. Nod1, an Apaf-1-like activator of caspase-9 and nuclear factor-kappaB. J.Biol.Chem. 1999, 274: 14560-14567.
- tructrucSuter T, Malipiero U, Otten L, Ludewig B, Muelethaler-Mottet A, Mach B, Reith W, Fontana A: Dendritic cells and differential usage of the MHC class II transactivator promoters in the central nervous system in experimental autoimmune encephalitis. Eur.J.Immunol. 2000, 30: 794-802.
-
*tructrucLandmann S, Muhlethaler-Mottet A, Bernasconi L, Suter T, Waldburger JM, Masternak K,Arrighi JF, Hauser C, Fontana A, Reith W: Maturation of dendritic cells is accompanied by rapid transcriptional silencing of class ii transactivator (ciita) expression. J.Exp.Med. 2001, 194: 379-392.
This paper represents a detailed analysis of the expression and promoter usage of the CIITA gene in immature DCs and during their maturation. -
**tructrucWaldburger JM, Suter T, Fontana A, Acha-Orbea H, Reith W: Selective abrogation of major histocompatibility complex class II expression on extrahematopoietic cells in mice lacking promoter IV of the class II transactivator gene. J.Exp.Med. 2001, 194: 393-406.
Mice lacking CIITA promoter IV exhibit selective abrogation of IFN-g induced MHCII expression on non-bone marrow-derived cells, but not on bone marrow-derived cells. Constitutive MHCII expression on cTECs, and thus positive selection of CD4+ T cells in the thymus are also abolished in these mice. - tructrucLennon AM, Ottone C, Rigaud G, Deaven LL, Longmire J, Fellous M, Bono R, Alcaide-Loridan C: Isolation of a B-cell-specific promoter for the human class II transactivator. Immunogenetics 1997, 45: 266-273.
- tructrucGoodwin BL, Xi H, Tejiram R, Eason DD, Ghosh N, Wright KL, Nagarajan U, Boss JM, Blanck G: Varying functions of specific major histocompatibility class II transactivator promoter III and IV elements in melanoma cell lines. Cell Growth Differ. 2001, 12: 327-335.
- tructrucDeffrennes V, Vedrenne J, Stolzenberg MC, Piskurich JF, Barbieri G, Ting JP, Charron D, Alcaide-Loridan C: Constitutive expression of MHC class II genes in melanoma cell lines results from the transcription of class II transactivator abnormally initiated from its B cell-specific promoter. J.Immunol. 2001, 167: 98-106.
- tructrucGhosh N, Piskurich JF, Wright G, Hassani K, Ting JP, Wright KL: A novel element and a TEF-2-like element activate the major histocompatibility complex class II transactivator in B-lymphocytes. J.Biol.Chem. 1999, 274: 32342-32350.
- tructrucPiskurich JF, Linhoff MW, Wang Y, Ting JP: Two distinct gamma interferon-inducible promoters of the major histocompatibility complex class II transactivator gene are differentially regulated by STAT1, interferon regulatory factor 1, and transforming growth factor beta. Mol.Cell Biol. 1999, 19: 431-440.
- tructrucKeller AD and Maniatis T: Identification and characterization of a novel repressor of beta- interferon gene expression. Genes Dev. 1991, 5: 868-879.
- tructrucKakkis E, Riggs KJ, Gillespie W, Calame K: A transcriptional repressor of c-myc. Nature 1989, 339: 718-721.
- *tructrucPiskurich JF, Lin KI, Lin Y, Wang Y, Ting JP, Calame K: BLIMP-I mediates extinction of major histocompatibility class II transactivator expression in plasma cells. Nat.Immunol. 2000, 1: 526-532.
-
*tructrucGhosh N, Gyory I, Wright G, Wood J, Wright KL: Positive regulatory domain I binding factor 1 silences class II transactivator expression in multiple myeloma cells. J.Biol.Chem. 2001, 276: 15264-15268.
MHCII expression is silenced during differentiation of B cells into plasma cells. Together with reference 130, this paper suggests that PRDI-BF1 / Blimp-1, a factor induced during differentiation into plasma cells, can repress expression of the CIITA gene. - tructrucTurner CA Jr., Mack DH, Davis MM: Blimp-1, a novel zinc finger-containing protein that can drive the maturation of B lymphocytes into immunoglobulin-secreting cells. Cell 1994, 77: 297-306.
- tructrucMuhlethaler-Mottet A, Di Berardino W, Otten LA, Mach B: Activation of the MHC class II transactivator CIITA by interferon-g requires cooperative interaction between Stat1 and USF-1. Immunity 1998, 8: 157-166.
- tructrucO'Keefe GM, Nguyen VT, Ping Tang LL, Benveniste EN: IFN-gamma regulation of class II transactivator promoter IV in macrophages and microglia: involvement of the suppressors of cytokine signaling-1 protein. J.Immunol. 2001, 166: 2260-2269.
- tructrucXi H, Eason DD, Ghosh D, Dovhey S, Wright KL, Blanck G: Co-occupancy of the interferon regulatory element of the class II transactivator (CIITA) type IV promoter by interferon regulatory factors 1 and 2. Oncogene 1999, 18: 5889-5903.
- tructrucXi H, Goodwin B, Shepherd AT, Blanck G: Impaired class II transactivator expression in mice lacking interferon regulatory factor-2. Oncogene 2001, 20: 4219-4227.
- tructrucPanek RB, Lee YJ, Benveniste EN: TGF-beta suppression of IFN-gamma-induced class II MHC gene expression does not involve inhibition of phosphorylation of JAK1, JAK2, or signal transducers and activators of transcription, or modification of IFN-gamma enhanced factor X expression. J.Immunol. 1995, 154: 610-619.
- tructrucDong Y, Tang L, Letterio JJ, Benveniste EN: The Smad3 protein is involved in TGF-beta inhibition of class II transactivator and class II MHC expression. J.Immunol. 2001, 167: 311-319.
- tructrucNavarrete SA, Kehlen A, Schutte W, Langner J, Riemann D: Regulation by transforming growth factor-beta1 of class II mRNA and protein expression in fibroblast-like synoviocytes from patients with rheumatoid arthritis. Int Immunol. 1998, 10: 601-607.
- tructrucEndo TA, Masuhara M, Yokouchi M, Suzuki R, Sakamoto H, Mitsui K, Matsumoto A, Tanimura S, Ohtsubo M, Misawa H, Miyazaki T, Leonor N, Taniguchi T, Fujita T, Kanakura Y, Komiya S, Yoshimura A: A new protein containing an SH2 domain that inhibits JAK kinases. Nature 1997, 387: 921-924.
- tructrucYasukawa H, Misawa H, Sakamoto H, Masuhara M, Sasaki A, Wakioka T, Ohtsuka S, Imaizumi T, Matsuda T, Ihle JN, Yoshimura A: The JAK-binding protein JAB inhibits Janus tyrosine kinase activity through binding in the activation loop. EMBO J. 1999, 18: 1309-1320.
- tructrucMorris AC, Spangler WE, Boss JM: Methylation of class II trans-activator promoter IV: a novel mechanism of MHC class II gene control. J.Immunol. 2000, 164: 4143-4149.
- tructrucvan den Elsen PJ, van der Stoep N, Vietor HE, Wilson L, van Zutphen M, Gobin SJ: Lack of CIITA expression is central to the absence of antigen presentation functions of trophoblast cells and is caused by methylation of the IFN-gamma inducible promoter (PIV) of CIITA. Hum.Immunol. 2000, 61: 850-862.
- tructrucAnderson G, Jenkinson EJ, Moore NC, Owen JJ: MHC class II-positive epithelium and mesenchyme cells are both required for T-cell development in the thymus. Nature 1993, 362: 70-73.
- tructrucHoney K and Rudensky A: The pIV-otal class II transactivator promoter regulates major histocompatibility complex class II expression in the thymus. J.Exp.Med. 2001, 194: F15-F18.
- tructrucErickson FL, Dinesh-Kumar SP, Holzberg S, Ustach CV, Dutton M, Handley V, Corr C, Baker BJ: Interactions between tobacco mosaic virus and the tobacco N gene. Philos.Trans.R.Soc.Lond.B.Biol.Sci. 1999, 354: 653-658.
-
**tructrucBeresford GW and Boss JM: CIITA coordinates multiple histone acetylation modifications at the HLA- DRA promoter. Nat.Immunol. 2001, 2: 652-657.
This in vivo study shows that acetylation and deacetylation of histone H3 and H4 at the HLA-DR promoter occurs with a time course that depends on CIITA expression and binding. - tructrucScholl T, Mahanta SK, Strominger JL: Specific complex formation between the type II bare lymphocyte syndrome- associated transactivators CIITA and RFX5. Proc.Natl.Acad.Sci.USA 1997, 94: 6330-6334.
- tructrucHake SB, Masternak K, Kammerbauer C, Janzen C, Reith W, Steimle V: CIITA leucine-rich repeats control nuclear localization, In vivo recruitment to the major histocompatibility complex (MHC) class II enhanceosome, and MHC class II gene transactivation. Mol.Cell Biol. 2000, 20: 7716-7725.
- tructrucRiley JL, Westerheide SD, Price JA, Brown JA, Boss JM: Activation of class II MHC genes requires both the X box and the class II transactivator (CIITA). Immunity 1995, 2: 533-543.
- tructrucZhou H and Glimcher LH: Human MHC class II gene transcription directed by the carboxyl terminus of CIITA, one of the defective genes in type II MHC combined immune deficiency. Immunity 1995, 2: 545-553.
- tructrucChin KC, Li GG, Ting JP: Importance of acidic, proline/serine/threonine-rich, and GTP- binding regions in the major histocompatibility complex class II transactivator: generation of transdominant-negative mutants. Proc.Natl.Acad.Sci.USA 1997, 94: 2501-2506.
- tructrucBontron S, Ucla C, Mach B, Steimle V: Efficient Repression of Endogenous Major Histocompatibility Complex Class II Expression through Dominant Negative CIITA Mutants Isolated by a Functional Selection Strategy. Mol.Cell.Biol. 1997, 17: 4249-4258.
- tructrucFontes JD, Jiang B, Peterlin BM: The class II trans- activator CIITA interacts with the TBP- associated factor TAF II 32. Nucleic Acids Res. 1997, 25: 2522-2528.
- tructrucYun S, Gustafsson K, Fabre JW: Suppression of MHC class II expression by human class II trans- activator constructs lacking the N-terminal domain. Int Immunol. 1997, 9: 1545-1553.
- tructrucZhou H, Su HS, Zhang X, Douhan J 3rd, Glimcher LH: CIITA-dependent and -independent class II MHC expression revealed by a dominant negative mutant. J.Immunol. 1997, 158: 4741-4749.
- tructrucMahanta SK, Scholl T, Yang FC, Strominger JL: Transactivation by CIITA, the type II bare lymphocyte syndrome- associated factor, requires participation of multiple regions of the TATA box binding protein. Proc.Natl.Acad.Sci.U.S.A. 1997, 94: 6324-6329.
- tructrucFontes JD, Kanazawa S, Jean D, Peterlin BM: Interactions between the class II transactivator and CREB binding protein increase transcription of major histocompatibility complex class II genes. Mol.Cell Biol. 1999, 19: 941-947.
- tructrucKanazawa S, Okamoto T, Peterlin BM: Tat competes with CIITA for the binding to P-TEFb and blocks the expression of MHC class II genes in HIV infection. Immunity 2000, 12: 61-70.
- tructrucSpilianakis C, Papamatheakis J, Kretsovali A: Acetylation by PCAF enhances CIITA nuclear accumulation and transactivation of major histocompatibility complex class II genes. Mol.Cell Biol. 2000, 20: 8489-8498.
- tructrucKretsovali A, Agalioti T, Spilianakis C, Tzortzakaki E, Merika M, Papamatheakis J: Involvement of CREB binding protein in expression of major histocompatibility complex class II genes via interaction with the class II transactivator. Mol.Cell Biol. 1998, 18: 6777-6783.
- tructrucHarton JA, Zika E, Ting JP: The histone acetyltransferase domains of CBP and pCAF are not necessary for cooperativity with CIITA. J.Biol.Chem. 2001, 276: 38715-38720
-
*tructrucZhu XS and Ting JP: A 36-Amino-Acid Region of CIITA Is an Effective Inhibitor of CBP: Novel Mechanism of Gamma Interferon-Mediated Suppression of Collagen alpha(2)(I) and Other Promoters. Mol.Cell Biol. 2001, 21: 7078-7088.
Together with references 179, 180 and 181, this paper suggests that CIITA may inhibit the expression of certain genes by sequestering the general coactivator CBP. - tructrucLee TI and Young RA: Transcription of eukaryotic protein-coding genes. Annu.Rev.Genet. 2000, 34: 77-137.
- tructrucStrahl BD and Allis CD: The language of covalent histone modifications. Nature 2000, 403: 41-45.
- tructrucRoth SY, Denu JM, Allis CD: Histone acetyltransferases. Annu.Rev.Biochem. 2001, 70: 81-120.
-
*tructrucRaval A, Howcroft TK, Weissman JD, Kirshner S, Zhu XS, Yokoyama K, Ting JP, Singer DS: Transcriptional coactivator, CIITA, is an acetyltransferase that bypasses a promoter requirement for TAF(II)250. Mol.Cell 2001, 7: 105-115.
This study suggests that CIITA has an endogenous acteyltransferase activity. - tructrucHarton JA, Cressman DE, Chin KC, Der CJ, Ting JP: GTP binding by class II transactivator: role in nuclear import. Science 1999, 285: 1402-1405.
- tructrucChin KC, Li G, Ting JP: Activation and transdominant suppression of MHC class II and HLA-DMB promoters by a series of C-terminal class II transactivator deletion mutants. J.Immunol. 1997, 159: 2789-2794.
- **tructrucKretsovali A, Spilianakis C, Dimakopoulos A, Makatounakis T, Papamatheakis J: Self-association of class II transactivator correlates with its intracellular localization and transactivation. J.Biol.Chem. 2001, 276: 32191-32197.
- **tructrucLinhoff MW, Harton JA, Cressman DE, Martin BK, Ting JP: Two distinct domains within CIITA mediate self-association: involvement of the GTP-binding and leucine-rich repeat domains. Mol.Cell Biol. 2001, 21: 3001-3011.
-
**tructrucSisk TJ, Roys S, Chang CH: Self-association of CIITA and its transactivation potential. Mol.Cell Biol. 2001, 21: 4919-4928.
**References 170 to 172 all demonstrate that CIITA requires self-association for its function, although there is some controversy concerning the precise domains that are involved. - tructrucCressman DE, Chin KC, Taxman DJ, Ting JP: A defect in the nuclear translocation of CIITA causes a form of type II bare lymphocyte syndrome. Immunity 1999, 10: 163-171.
- tructrucCressman DE, O'Connor WJ, Greer SF, Zhu XS, Ting JP: Mechanisms of nuclear import and export that control the subcellular localization of class ii transactivator. J.Immunol. 2001, 167: 3626-3634.
- tructrucTowey M and Kelly AP: Nuclear localisation of CIITA is controlled by a carboxy terminal leucine-rich repeat region. Mol.Immunol. 2002, 38: 627-634.
- tructrucGobin SJ, Peijnenburg A, Keijsers V, van den Elsen PJ: Site alpha is crucial for two routes of IFN gamma-induced MHC class I transactivation: the ISRE-mediated route and a novel pathway involving CIITA. Immunity 1997, 6: 601-611.
- tructrucMartin BK, Chin KC, Olsen JC, Skinner CA, Dey A, Ozato K, Ting JP: Induction of MHC class I expression by the MHC class II transactivator CIITA. Immunity 1997, 6: 591-600.
- tructrucGobin SJ, Peijnenburg A, Van Eggermond M, van Zutphen M, van den Berg R, van den Elsen PJ: The RFX complex is crucial for the constitutive and CIITA-mediated transactivation of MHC class I and beta2-microglobulin genes. Immunity 1998, 9: 531-541.
- tructrucGourley T, Roys S, Lukacs NW, Kunkel SL, Flavell RA, Chang CH: A novel role for the major histocompatibility complex class II transactivator CIITA in the repression of IL-4 production. Immunity 1999, 10: 377-386.
- tructrucGourley TS and Chang CH: Cutting edge: the class II transactivator prevents activation-induced cell death by inhibiting Fas ligand gene expression. J.Immunol. 2001, 166: 2917-2921.
- tructrucSisk TJ, Gourley T, Roys S, Chang CH: MHC class II transactivator inhibits IL-4 gene transcription by competing with NF-AT to bind the coactivator CREB binding protein (CBP)/p300. J.Immunol. 2000, 165: 2511-2517.
- tructrucSaifuddin M, Roebuck KA, Chang C, Ting JP, Spear GT: Cutting edge: activation of HIV-1 transcription by the MHC class II transactivator. J.Immunol. 2000, 164: 3941-3945.
- tructrucSaifuddin M, Spear GT, Chang C, Roebuck KA: Expression of MHC class II in T cells is associated with increased HIV-1 expression. Clin.Exp.Immunol. 2000, 121: 324-331.
- tructrucOkamoto H, Asamitsu K, Nishimura H, Kamatani N, Okamoto T: Reciprocal modulation of transcriptional activities between HIV-1 Tat and MHC class II transactivator CIITA. Biochem.Biophys.Res.Commun. 2000, 279 :494-499.
-
**tructrucAccolla RS, De Lerma BA, Mazza A, Casoli C, De Maria A, Tosi G: The MHC class II transactivator: prey and hunter in infectious diseases. Trends Immunol. 2001, 22: 560-563.
This review discusses a series of results (references 159, 182, 184 and 186) addressing the interaction between CIITA and pathogens such as HIV. - tructrucTosi G, De Lerma BA, D'Agostino A, Valle MT, Megiovanni AM, Manca F, Caputo A, Barbanti-Brodano G, Accolla RS: HIV-1 Tat mutants in the cysteine-rich region downregulate HLA class II expression in T lymphocytic and macrophage cell lines. Eur.J.Immunol. 2000, 30: 19-28.
- tructrucMudhasani R and Fontes JD: Inhibition of class II trans-activator function by HIV-1 tat in mouse cells is independent of competition for binding to cyclin T1. Mol.Immunol. 2002, 38: 539-546.
- tructrucLe Roy E, Muhlethaler-Mottet A, Davrinche C, Mach B, Davignon JL: Escape of human cytomegalovirus from HLA-DR-restricted CD4(+) T-cell response is mediated by repression of gamma interferon-induced class II transactivator expression. J.Virol. 1999, 73: 6582-6589.
2.3 Dendritic cells
2.3.1 The role of dendritic cells in the immune system
Host defense relies on a concerted action of both antigen-nonspecific innate immunity and antigen-specific adaptive immunity (140, 141). Key features of the innate immune system include the ability to rapidly recognize pathogens and/or tissue injury and the ability to signal the presence of danger to cells of the adaptive immune system (142). The innate system includes phagocytic cells, natural killer (NK) cells, complement and interferons (IFNs). Cells of the innate system use a variety of germline-encoded pattern recognition receptors to recognize patterns shared between pathogens, for instance bacterial LPS, carbohydrates, and double-stranded viral RNA (143-145). Evolutionary pressure has led to the development of adaptive immunity, the key features of which are the ability to rearrange genes of the immunoglobulin family, permitting creation of a large diversity of antigen-specific clones and immunological memory. Yet this highly sophisticated and potent system needs to be instructed and regulated by APCs. DCs are unique APCs because they are the only ones that are able to induce primary immune responses, thus permitting establishment of immunological memory (98-100).
DCs represent a heterogeneous cell population, residing in most peripheral tissues, particularly at sites of interface with the environment (skin, mucosae) (98). They display a high phagocytic capacity. Following tissue damage, DCs process the captured antigens, load them on MHC molecules and migrate to the lymphoid organs, where they present the antigenic peptides in the context of MHC molecules to T cells, thereby initiating adaptive immune responses (146, 147). DCs activate antigen-specific CD4+ T helper cells, which in turn regulate the immune effectors, including antigen-specific CD8+ cytotoxic T cells (CTLs) and B cells, as well as non-antigen-specific macrophages, eosinophils and NK cells. Moreover, DCs educate effector cells to home to the site of injury (98). Four stages of DC development have been delineated, including 1) bone marrow progenitors; 2) precursor DCs that are patrolling through blood and lymphatics as well as lymphoid tissues, and that upon pathogen recognition, release large amounts of cytokines, e.g. IFN-a, thereby limiting the spread of infection; 3) tissue-residing immature DCs, which possess high endocytic and phagocytic capacity permitting antigen capture; and 4) mature DCs, present within secondary lymphoid organs, that express high levels of costimulatory molecules permitting antigen presentation (Fig.11).
Circulating precursor DCs enter tissues as immature DCs. They may directly encounter pathogens that induce secretion of cytokines (e.g. IFN-a). Immature DCs reside at strategically important sites in the periphery to encounter pathogens. After antigen capture, immature DCs migrate to lymphoid organs where, after maturation, they display peptide-MHC complexes, which allows selection of rare circulating antigen-specific lymphocytes. These activated T cells help DCs in terminal maturation, which allows lymphocyte expansion and differentiation. Activated T lymphocytes migrate and reach the injured tissues. Helper T cells secrete cytokines, which permit activation of macrophages, NK cells and eosinophils. Cytotoxic T cells eventually lyse the infected cells. B cells become activated after contact with T cells and DCs and then mature into antibody-producing plasma cells. It is believed that, after interaction with lymphocytes, DCs die by apoptosis.
2.3.2 Immature dendritic cells
Immature DCs reside in peripheral tissues at sentinel positions where they sample self and non-self antigens. However, they are not able to present the antigens. Immature DCs accumulate MHC molecules intracellularly and present only a small fraction at the cell surface (103, 104).
Three types of antigen uptake are known: macropinocytosis, phagocytosis and clathrin-mediated endocytosis (101, 102). Different types of antigens are internalized via these different routes. The constitutive and cytoskeleton-dependent process of macropinocytosis allows rapid and non-specific sampling of large amounts of surrounding fluid and results in the formation of large intracellular vacuoles. Phagocytosis is a receptor-mediated process dependent on regulated actin assembly. In general, the same receptors mediating phagocytosis of pathogens are also engaged in clathrin-dependent endocytosis. The latter allows uptake of macromolecules through specialized regions of the plasma membrane, termed coated pits (148). A large number of endocytic receptors are expressed on immature DCs, namely C-type lectins (101, 149, 150), receptors for the Fc portion of immunoglobulins (FcRs) (151, 152), complement receptors (153), receptors for heat shock proteins (154, 155), and scavenger receptors (156).
2.3.3 Dendritic cell activation and maturation
A signal from pathogens, often referred to as a danger signal (157), induces DCs to enter a developmental program, called maturation, which transforms DCs from sentinels into efficient APCs and T cell stimulators (98). The danger signal can be a bacterial or viral product as well as an inflammatory cytokine. Danger signals are recognized through specific pattern-recognition receptors, such as Toll-like receptors, FcRs and cytokine receptors (151, 158).
With the induction of maturation, DCs undergo massive morphological and functional changes. Antigen internalization is downmodulated due to repression of most antigen-receptors and to an overall reduction in phagocytosis and macropinocytosis (101). In addition, MHC molecules are redistributed from intracellular endocytic compartments to the cell surface (103, 104). Peptide loading as well as the half-life of MHC molecules is increased (103, 104). Finally, the surface expression of T cell costimulatory molecules also rises.
Concomitant with the modifications of their antigen presentation abilities, maturation induces migration of DCs out of peripheral tissues (105). Modifications in the expression of chemokine receptors and adhesion molecules, as well as profound changes of the cytoskeleton organization, contribute to the migration of DCs, through the lymph, towards secondary lymphoid organs (106). By linking antigen uptake, peptide loading, and cell migration to the encounter of a danger signal, DCs restrict antigen presentation to those antigens internalized during maturation, thus favoring the stimulation of T cells specific for potentially pathogenic antigens.
2.3.4 Antigen presentation by dendritic cells
CD8+ and CD4+ T cells express clonally distributed TCRs that recognize antigenic peptides associated with MHCI and MHCII molecules, respectively. A strict compartmentalization of MHCI and MHCII biogenesis results in the loading of exogenous antigens on MHCII molecules in the endocytic pathway, and the selective loading of endogenous antigens on MHCI molecules in the ER. This model accounts for the selective killing by MHCI-restricted CD8+ T cells of virus-infected cells (expressing endogenous viral antigens), but not of neighboring cells that have internalized inactive virus or apoptotic cells.
Peptides to be loaded onto MHCI molecules are generated by proteasome-mediated degradation of newly synthesized ubiquitinylated proteins (Fig.12). The immunoproteasome, which is expressed at low levels in immature DCs, becomes the main type of proteasome in mature DCs (107-109). The peptides resulting from proteasome mediated degradation are transferred to the ER by a specialized peptide transporter, TAP, and loaded onto newly synthesized MHCI molecules under the control of several ER resident chaperones (159). MHCI-peptide complexes are then rapidly transferred through the Golgi apparatus to the plasma membrane. MHCI synthesis and half-life are slightly increased during DC maturation (103, 104).
Fig. 12 MHCI restricted antigen presentation.

Pathogen-derived or self-proteins within the cytosol of APCs are enzymatically digested into peptides, mainly by the cytosolic protease called proteasome, and are then transported by TAP molecules into the ER. In the ER lumen, peptides bind to MHCI molecules, which are subsequently transported via the Golgi to the plasma membrane.
The formation of MHCII-peptide complexes in DCs is induced by maturation. This is different from the situation in other APCs, where this process is constitutive. In immature DCs, most MHCII molecules are retained in late-endosomal and lysosomal compartments (Fig.13). They are persistently associated with the invariant chain Ii, which is poorly degraded in these cells (110, 111) and thus prevents peptide loading. However, upon induction of maturation, Ii is degraded by cathepsin S and MHCII-peptide complexes are formed and transported to the surface (110). Additional mechanisms for the regulation of MHCII-peptide complex formation and presentation exist in DCs. First, internalized antigens are processed into peptides only once the cell starts to mature (112). Second, de novo MHCII synthesis is transiently upregulated during DC maturation (103, 104). Finally, the reduction in endocytosis slows down the recycling of the MHCII molecules at the cell surface and thus contributes to their increased half-life.
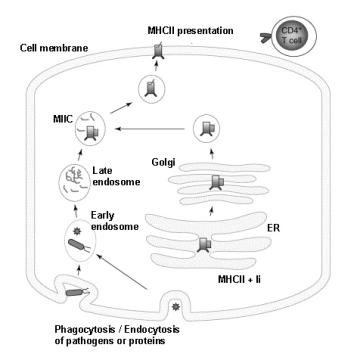
Exogenous antigens are derived from proteins that are endocytosed and processed by proteases. Newly synthesized MHCII molecules are retained in lysosomal compartments. Peptide loading occurs upon DC maturation in specialized antigen-processing vesicles, MIIC. The peptide-MHCII complexes are then externalized to the plasma membrane.
In contrast to the model discussed above, exogenous antigens can also be presented by MHCI molecules, thereby leading to CD8+ T cell activation (114-116). This phenomenon is called cross-presentation. It is crucial for the initiation of cytotoxic immune responses against antigens that are not synthesized by the DCs themselves, but by other cells. Normally, antigen-bearing cells do not directly initiate immune responses, since they are not capable of stimulating resting naive T cells. Most immune responses thus require antigen transfer to professional APCs, most probably DCs (160, 161), which then stimulate naive T cells. Apoptotic cells seem to be favorite targets for cross-presentation (161-163). Two intracellular pathways can result in cross-presentation: TAP-independent endocytotic or TAP-dependent ER MHCI peptide loading (164) (Fig.14). The latter implicates the existence of an endosome-to-cytosol transport system (165).

a) Exogenous antigens can access MHCI molecules within endosomes that contain MHCI molecules that have been recycled from the membrane. b) Alternatively, antigen is transported from the endosome to the cytosol and processed similarly to endogenously derived antigens.
2.3.5 T cell stimulation and tolerization by dendritic cells
DCs have the unique ability to prime naïve CD4+ and CD8+ T cells. This has been shown in in vivo, in vitro and in situ experiments (166-175). In most cases, CD4+ T cell help is required for efficient CTL activation. In traditional models of CTL activation, the CD4+ and CD8+ T cells were thought to recognize antigen on the same APC. However, in the current model (176-178), the APCs are licensed to activate CTLs by T helper cells via upregulation of CD40 ligand (CD40L) on the DCs. Thus, a conditioned DC becomes a temporal bridge between a CD4+ T helper cell and a CTL. In addition to priming, DCs appear essential to maintain survival of naive CD4+ T cells (123) and immune T cell memory (179). Importantly, DCs are also involved in the tolerization of the T cell repertoire to self-antigen. This occurs in the thymus (central tolerance) by deletion of developing T cells (123), and in lymphoid organs (peripheral tolerance) probably by the induction of anergy or deletion of mature T cells.
DCs play apparently contradictory roles by stimulating as well as tolerizing T cells. This inconsistency may be attributed either to distinct DC lineages endowed with unique T cell stimulatory capacity or to a single DC type, which is instructed by environmental stimuli to perform different functions. Indeed, two major subtypes of DCs, that exhibit different functions, exist: myeloid and lymphoid (98). In addition, the different DC functions can be altered by the cytokine environment.