[Previous]
1. Introduction
Dentists may be confronted with patients suffering from a wide range of systemic disorders. Some of these illnesses can have primary or secondary manifestations in the face and the oral cavity. It is therefore important to have a basic knowledge about these manifestations, in order to recognize them as a part of a general condition and bring them to adequate medical attention. In certain cases the first signs of illness are in or around the oral mucosa. Once recognized as a pathological lesion the dentist can play an important role in referring the patient for a check up which hopefully will lead to a quicker and more effective treatment of the disease at an early stage.
Scleroderma is a multi-system disease, within the group of connective tissue diseases. Scleroderma is of unknown etiology, characterized by abnormal synthesis of connective tissues. In most cases of systemic sclerosis, vascular and fibrotic changes cause fibrosis (scarring) of the skin and even of internal organs at various degrees. It also exists in a localized form.
The intense sclerosis of organs and primarily the fibrosis of the skin has devastating effects on the affected patients on several levels. In the mouth and the facial region bone resorption and hampering of the normal mouth aperture with associated nutrition and hygiene problems can have widespread effects on both the oral mucosa and the dentition. The obvious problem to provide adequate dental care to patients with limited mouth aperture makes it even more important to detect the very first signs of illness to prevent dental and oral illness with prophylactic measures.
This thesis is a literature review that outlines the oral manifestations and thus the given dilemmas of scleroderma.
2. Terminology
In systemic sclerosis, the most apparent signs of disease are manifested in the skin and it is to these signs that the disorder owes it's original name scleroderma. Scleroderma (Gr. skleros, hard, and derma, skin) is a generic term, used to describe both systemic as well as more localized cutaneous disorders. When the systemic nature of the disease became evident, the name progressive systemic sclerosis was proposed (30). Subsequently the term progressive was removed, mainly because of the psychological impact of the diagnosis on the patients.
The most frequent diagnostic criteria used for scientific studies (28,76) of this condition employ the term systemic sclerosis, which is accordingly the one used in this thesis.
In practical clinical terms, the word scleroderma has been retained to refer to both the disease, which is here called systemic sclerosis (SSc), and for the other conditions within the heterogeneous group of diseases, which involve the hardening, tightening and decreasing elasticity of the skin (71). Thus scleroderma represents from a clinical point of view, a spectrum of different disease entities ranging from localized scleroderma to overlap syndromes with mainly systemic manifestations (Tab 1a,1b); overlap syndromes are in this context, used when characteristics of scleroderma are associated with characteristics of Sjögren's syndrome, systemic lupus erythemathosus, rheumatoid arthritis, dermato -or polymyositis and early mixed connective tissue disease.
Systemic sclerosis can, in turn, be subdivided into conditions characterized either mainly by diffuse skin involvement affecting both the trunk, the extremities and the face and into a more limited form, which was historically called acrosclerosis (acroscleroderma) but which is now generally called the CREST syndrome (Calcinosis, Raynaud's phenomenon, Esophaghypomotility, Sclerodactyly and Telangiectasia) or the limited cutanous form of SSc (45).
Scleroderma limited to the skin only can be subdivided into morphea and linear scleroderma.
 Table 1a. The clinical spectrum of scleroderma. In literature the concept of scleroderma is synonymous with all subgroups presented above. Schema adapted from Gustavsson et al (32) .
Table 1a. The clinical spectrum of scleroderma. In literature the concept of scleroderma is synonymous with all subgroups presented above. Schema adapted from Gustavsson et al (32) .
| Table 1b. Classification according to American Rheumatism Association (88) and others. |
| A. Criteria for scleroderma |
| American Rheumatism Association classification |
Major criteria: proximal scleroderma: scleroderma involvement proximal to the digits |
| Minor criteria: sclerodactyly (scleroderma involvement in the finger and toes), pulmonary fibrosis and digital pitting scars |
| B. Subset of systemic scleosis |
| Barnett (1) |
Three types (I,II,III) according to early-stage skin involvement |
| Giordano (28) |
Sclerosis sine scleroderma, limited, intermediate and diffuse cutaneous sclerosis |
| LeRoy et al (45) |
Limited and diffuse cutaneous form |
| Rodnan et al (76) |
CREST syndrome, diffuse scleroderma and overlap syndrome |
| Winkelmann (96) |
Vascular scleroderma and inflammatory scleroderma |
3. General introduction to systemic sclerosis (scleroderma)
Systemic sclerosis, as a term is characterized by intense fibrosis. The skin, blood vessels, synovium and skeletal muscles along with the following internal organs, gastrointestinal tract, lung, heart and kidney can be involved at different levels.
Clinically, in most of it's forms, SSc is characterized by induration, loss of the skin contours and the skins ability to wrinkle, and immobility over underlying tissues. The skin thickening always starts in the fingers (84) .
There is a broad spectrum of combinations and severity of the organs affected. The main difference between the groups (localized scleroderma and systemic sclerosis) is the pace at which the disease progresses and the number of organs involved.
Localized scleroderma patients can show edematous (swollen, fluffy) fingers for years without actually getting the skin thickening. The skin thickening may extend to the hands and distal forearms as well as neck and trunk but the evolution to systemic sclerosis is rare. Patients with diffuse systemic sclerosis may also show fluffy fingers, Raynaud´s phenomenon (an episodic change in color of the fingertips in response to cold or emotions), similar to patients with localized scleroderma, but are likely to have a faster progression of other manifestations than patients with the localized form (107) . The diagnosis is always done by examination of the patients hands.
The diagnostic criteria's according to the American Rheumatism Association of SSc are: Either scarring ("scleroderma") of the skin in the extremities, or two of the following symptoms in the same patient; sclerodactily, edematous lesions of the fingers, loss of sensitivity in the fingertips or bilateral basiliar pulmonary fibrosis on chest roentgenogram (32,86).
4. Historical background
Hippocrates (460-377 BC) was the first to describe a skin disorder compatible to scleroderma but due to a rather imprecise description this report can not be relied upon (73) .The first convincing description of the disease appeared in a monography written by Carlo Curizo, in Naples in 1753. He reported on the hardness in the skin of a 17 year old woman (15) . SSc was first given the name sclérodermie by Gintrac in 1847 (27) . However it was not until 1924 when Matsuis description of five patients with sclerodermie involving the lungs, kidneys and gastrointestinal tract, that the visceral involvement was recognized (52) .
Goetz (30) proposed the name progressive systemic sclerosis in 1945, when the systemic nature of the disease was proven. The name has though been replaced by systemic sclerosis, since not all cases progress, and also to diminish the psychological impact of the diagnosis (44).
5. Epidemiology and prognosis
Systemic sclerosis exists geographically over the entire globe.
Epidemiological studies have been hampered by its relative rarity. The annual incidence of SSc is reported to be about 4,5-14,5 new cases per 1 million people. In the U.S only an estimated 50.000-100.000 people (26, 56, 58,63,101) are diagnosed with scleroderma. Other reasons for hampering further epidemiological research is the lack of specific or sensitive diagnostic laboratory tests and the tendency to share clinical features with other connective tissue diseases.
SSc rarely develops in children, the onset is generally in the middle aged adult, 30-50 years of age (26,30) . The ratio of female to male patients is approximately 3 to 1, and peaks before menopause (26,30,44,57) .
All races are affected. There is a slight difference in the rate of occurrence between blacks and whites, with the risk for blacks being greater than for whites (71) . The survival of patients with systemic sclerosis has been examined in large retrospective studies (30,75,89) . The five year survival rate has ranged from 34% to 73%. One reason for the variability of reported survival rates may be the difficulties involved in assigning the patients into prognostically usable subgroups (30) , an early detection and assigning of patients with an early systemic sclerosis according to clinical features and serological characteristics provides a reasonable prognostic indication. Several other factors influence the survival rate, the numbers of organs involved and the severity of the disease seem to play an important role. The severity of the disease can often be determined during the first two years of illness. Another factor is age (2,94) . Renal, cardiac and pulmonary involvement changes the prognostic to the worse (8,90) .
Renal involvement has the worst prognostic value followed by cardiac and pulmonary involvement. There is now however, evidence suggesting that early aggressive therapy of renal failure, in particular with ACE-inhibitors, may improve the survival rate.
6. Etiology and pathogenesis
The familial predilections are not clear, but auto-antigens are detected in parents of patients with SSc suggesting a hereditary predisposition (75,87,90) .
Several risk factors have been proposed. For example, in coal or gold mines workers, and other occupationally exposed to silica dust (26,28,34,42,96,101) . People working with polyvinyl and paraffin or silicone implantation for breast augmentation purposes (40,41,63) are also thought of being at higher risk and are said to develop Raynaud´s phenomenon, acro-osteolysis, cutaneous lesions which can be called scleroderma-like illnesses, this is subject to discussion and has not been proven.
Various hypotheses to explain the etiology of SSc are proposed; infectious, biomechanical, immunological, and vascular, but the disease is still of unknown origin (26,81) . Tab. 2 summarizes the different hypothesis.
| Table 2. Pathogenic factors reproduced from Jaworsky (46). |
| Vascular |
Raynaud`s phenomenon |
| Aberrations |
Intimal hyperplasia of blood vessels |
| |
Adventitial fibrosis |
| |
Vascular abnormalities in other visceral organs |
| |
Microvascular abnormalities |
| |
Enlargement and tortuosity of capillary loops |
| |
Capillary loop dropout |
| |
Thickened and reduplicated basement membrane |
| |
Loss of endothelium |
| |
|
| Immunologic |
T helper cell infiltrates |
| Factors |
Reduction in T suppressor cells |
| |
Increased soluble interleukin 2 receptors correlate with |
| |
disease progression and mortality |
| |
|
| Serologic |
ANA+ in more than 90% of patients |
| Factors |
Low/absence of anti-native (double stranded) DNA, anti-Sm, rare anti-nRNP |
| Speckled ANA pattern correlates with anti-centromere antibody on Hep-2 cells and CREST and a relatively |
| favorable prognosis |
| Anti-DNA-Topoisomerase 1 |
6.1 Environmental causative factor theory
In 1914, a Scottish physician, Bramwell (6,14,18,72,) reported on nine diagnosed scleroderma patients, of which five where working as stone masons. Bramwell´s work suggested that holding the chisel and working in the open air under cold-weather conditions were work related causative factors.
In 1957, Erasmus (18) encountered 17 cases of systemic sclerosis among 8000 underground miners during a period of 18 months. This and other discoveries has shown an elevated prevalence of SSc in mine workers, since 1974 SSc in miners is acknowledged as an occupational disease that has to be compensated by law. An association between SSc and silicosis has been described more than once in the medical literature (17,24,70,80,92) . The report on the obviously increased prevalence of SSc in workers exposed to silica dust, mainly in the mining of industry in the German Democratic Republic was published 1976 by Zschunke (101) . A first analysis, carried out in the whole country, was performed in 1981 and showed that 77 percent of all male patients with SSc were exposed to silica dust in their work place.
It has been discussed whether this is true SSc or should be classified as scleroderma like syndrome (101).
6.2 Vascular theory
The vascular theory tries to explain the etiology of SSc as follows. In SSc patients there is always vascular changes. The vascular changes can be subdivided into two, namely, vasomotor instability and small vessel structural changes. The most obvious proof of an existing vasomotor instability in patients with SSc is the universal prevalence of Reynaud´s syndrome (7,16,25,43,) , which is the initial symptom of SSc. The cause of Raynaud´s phenomenon is unknown. There are contradictory results published from studies concerning the relation between the severity of Reynaud's phenomenon and visceral manifestations of SSc. Pathology in the large arteries are very rare, but when it occurs it can secondarily lead to ischemic damage in the larger vessels (25) . Lesions in small arteries and arterioles are on the contrary found in almost all organs. Typical histopathological changes in these small arteries include intimal thickening with mucoid infiltration, medial thinning, periadventitial fibrosis and a large amount of fibrin deposition. Abnormalities in capillaries in patients with SSc include typical scleroderma pattern on nail fold capillary with an adjacent vascular area in 82% of the SSc patients (50) . Injury to the endothelial cells often leads to platelet aggregation and since platelets contain a large number of active mediators for the induction of fibroblast function, endothelial cell injury can represent a key event in the development of fibrosis in SSc.
The mechanism behind this endothelial cell injury is still unknown but a theory is that specific factors seem to be toxic to the cells (13,34) .
Other explanation of this injury to the endothelial cells can be an exogenous stimulus such as disposition of immune complexes (53,67) and immunoglobulin complement factors and fibrin (42) .
6.3 Immune system theory
There is numerous indirect evidence that immunological mechanisms are involved in the pathogenesis of SSc. First, there is a strong association with other disorders connected with immunological alterations such as Sjögren's syndrome, lupus erythematosus and polymyositis (44) . Secondly the presence of auto-antibodies directed against nuclear and cellular antigens is yet another indirect evidence. Although some of these antibodies are highly specific for SSc, there is still no evidence that they are involved in the pathogenesis or play an active role in the cause of the disease (9) .
A more direct proof of immunological involvement is the appearance of scleroderma like lesions in patients with known purely immunological disease's such as seen in chronic graft-versus-host disease, secondary to bone marrow transplantation (85) . Several immunological abnormalities are seen in patients with SSc. Antibody factors are found in 95 out of 100 cases and a hypergammagloubulin in at the least half of the patients (22,38,48) The immunogenetic studies to demonstrate associations with one more HLA antigens have been inconclusive(22,38) . Positive titers of serum antinuclear antibodies (ANA) are often found in the sera of SSc patients. Depending of the used substrate frequency ranges greatly among different research groups. One has identified at least two relatively specific antibodies. These are the anti-Scl-70 and the anticentromere antibody (ACA). The anti-Scl-70 is associated with patients with diffuse systemic sclerosis whereas (ACA) is closer connected to the CREST syndrome. ACA is found almost only in patients with the CREST syndrome but less than 50% of the CREST patients have the ACA antibody. No patient has been found to have both. One third of the SSc patients presents presence of Rheumatoid factors at low titers.
The latest research states that calcinosis generally results from dystrophic calcification associated with massive collagen degeneration. However, the pathogenesis of calcinosis is not well understood. Transforming growth factorß (TGFß) is known to play an important role of regulation extracellular matrix (ECM) in dermis metabolism and increasing collagen production by fibroblasts. TGFß may also play a critical role in SSc and is a potential contributor to developing SSc. TGFß has been hypothesied as the link between the inflammatory phase and the subsequent fibrotic reaction of SSc. Their findings demonstrate a close association between the TGF ß3 protein and the fibroblastic and inflammatory cell populations in SSc which supports an aetiological role of TGF ß 3 in the calcifiying process of SSc. Similarily the vasospasm associated with Raynaud's phenomenon has been shown to reduce peripheral tissue oxygen levels, hypoxia has been shown to augment TGFß3 secretion from endothelial cells and potentiate the fibrotic effect of TGF (36).
7. Localized scleroderma
Localized scleroderma can be subdivided into two main forms Tab 3 (57) .
 Table 3. The clinical spectrum of localized scleroderma
Table 3. The clinical spectrum of localized scleroderma
7.1 Clinical features
These lesions are usually limited to the skin and subcutaneous tissue beneath the cutaneous lesions, but occasionally the underlying muscles are also affected. Although morphea may be associated with arthralgia (pain in joints and during articulation without detectable joint lesions)and in most cases with Raynaud's phenomenon (an episodic change in color of the fingertips in response to cold or emotions) generally accepted instances of transition from morphea to systemic sclerosis with visceral involvement has been reported as very rare (20) .
Lesion of morphea type shows a pale indurated plaques with smooth surface and internal sclerosis. Many plaques may be surrounded by a violaceous halo (ring). The prognosis is usually good and the fibrosis slowly clears leaving slight depression and hyperpigmentation. A rare type may lead to arrest of growth of underlying bones causing, for example, facial hemiatrophy (Romberg's disease) or shortening of limb. 95% of these patiens show a presence of Raynaud's phenomenon (7,16,24,43,63), which is characterized by digital cyanosis and blanching that results from internal hyperplasia of the digital arteries causing structural narrowing. The presence of vascular changes can be seen with widefield microscopy of the nailfold capillary bed (50,97) .The finding of enlarged, dilated tourtus capillary loops with capillary dropout is typical for scleroderma.
Lesions of the linear type occur predominantly on the extremities and on the anterior scalp. It also can occur in the face, where it results in hemiathrophy, on one or several extremities, where it produces in addition to induration of the skin, marked atrophy of the subcutaneous fat and of the muscles and tendons, and ankyloses of the joints at the hands and feet. On the anterior portion of the scalp the linear lesions often shows the configuration of the stroke of a saber ("coup de sabre") (81).
7.2 Localized scleroderma histopathology
Scleroderma is diagnosed clinically. Histological changes are present. It is important that the specimen biopsy include adequate amount of subcutaneous tissue, since most of the diagnostic alterations are observed there (81).
Two stages can be detected: an early inflammatory and a late sclerotic stage.
In the early inflammatory stage, found particularly at the violacous border of enlarging lesions, the reticular dermis shows closely packed, thick collagen bundles and a fairly mild inflammatory infiltrate, predominantly lymphocythic, between the collagen bundles and around the blood vessels (91) .
A much more pronounced inflammatory infiltrate than that seen in the dermis involves the subcutaneous fat and its upward projections toward the eccrine glands. In the late sclerotic stage, as in the center of a lesion, the inflammatory infiltrate has disappeared, except for in some areas of the subcutis. The epidermis is normal. The collagen bundles are thickened, packed tighter and show hypocellularity that stains eosinophilic more than in healthy skin. The eccrine glands appear markedly atrophic and are surrounded by newly formed collagen, they also seem to lie higher in the dermis than normal. This because of that the subcutaneous fat has been replaced by newly formed collagen. Very few blood vessels are noted here, they often have fibrotic walls with narrowed lumen. The septa subdividing the subcutaneous fat are thickened because of the presence of an inflammatory infiltrate and the deposition of new collagen.
Large areas of subcutaneous fat are being replaced by newly formed collagen that is composed of fibers rather than of bundles and strains only. Vascular changes are mild in the dermis and subcutaneous tissue during the early inflammatory stage: these changes can constitute of endothelial swellings and edema of the vessels walls.
Even the fascias of the striated muscles can show signs of fibrosis. The muscle fibers then appear vacuolated and separated from one another by edema and focal collection of inflammatory cells
The established lesion in the epidermis is normal or shows variable degree of atrophy and occasionally acanthosis, hypopigmentation or hyperpigmentation of the basal layer of the epidermis.
In a later stag, the collagen bundles of the corium become brad and sclerotic the number of the nuclei are diminished and fibrotic tissue proliferated and the extension into the subcutaneous fat resulting in the thickening of the entire cutis.
7.3 Localized scleroderma differential diagnosis
The dyschromes which are seen together with the morpheas are sometimes clinically dominating. This can cause confusion in the diagnostics with vitiligo (white spots well contoured in the skin) or a Becker naevus (brown spots on the skin with hairs coming out). These dermatoses can coexist at the same time in the same patient
7.4 Localized scleroderma therapy
The treatment is symptomatic. Each affect treated separately.
8. Systemic sclerosis
8.1 Clinical features
The onset is usually faster than the one seen in patients with localized scleroderma. The patient is a female in the age of 30-50. The pace at which the disease progresses is usually faster as well, 75% or more of these patients show the presence of Raynaud's phenomenon and it is often the first symptom of SSc. Raynaud's phenomenon can precede the disease from a couple of weeks until a couple of years. A short interval between the first signs of Raynaud´s phenomenon until the first signs of sclerosis is a bad prognostic sign. The first signs for the patients is often when the hands and face become fluffy (edematous) and stiff in the morning (86) . On examination, the findings of skin puffy or taut is normal. Further investigations show in most cases esophageal hypomotility and restrictive pulmonary abnormalities. However SSc may involve all connective tissue organs, though some represents main targets of the disease such as the skin, the gastrointestinal tract, the heart, the lungs and the kidneys.
8.1.1 Skin involvement
As mentioned above skin thickening in SSc will always begin in the fingers. If a patient has significant thickening of skin on the forearms, trunk, or legs without involvement of the fingers it is likely a scleroderma like disorder. This edematous phase lasts for a variable amount of time and can easily be confused with severe synovitis (26) . At some time this puffiness turns into skin thickening, which progresses up the extremities and then involves the trunk. Most progression of skin thickening occurs in the first 3 or 4 years of the illness. The diagnostic means of Reynaud's phenomenon is purely clinical (86) , initiated by cold it starts with a brutal drop (syncopale), one or several fingers turn white , cold and insensitive. This phase then changes into a phase of suffocation, shortness of air (asphyxia) within only a couple of minutes (63,86) . The fingers turn cyanotic, (blue in colors due to the lowered oxygen uptake of the hemoglobin) and painful, then finally red as recovery occurs. The duration of the crises varies from patient to patient. With time these crises of Reynaud's phenomenon are easier started and sclerodactylie (sclerosis of the fingers) takes its place. Rapid development of hand swelling and skin tightening often lead to severe flexion contractors with claw like hands as a result, giving deformities and serious disability. The skin gets mummified, the face expressionless. The mimic gestures are wiped away and displaced by a smooth leather like skin. Subcutaneous calcinosis occurs in about 20% of the SSc patients, it is related to disease duration and increases with time. This calcinosis defines the syndrome of Thibierge and Weissenbach, later called the CREST syndrome.
This syndrome is essentially seen on the legs and fingers, forming nodules or palpable or even visible masses. This is due to the local ischemic conditions. Ulcerations can occur secondary to vascular insufficiency, subcutaneous calcifications. These ulcerations display no healing and are very painful to the patient.
Other skin manifestations include pruritus, skin dryness, hair loss, pigment changes and telangiectasias. Telangiectasia has been successfully treated with surgical laser, but they are likely to reoccur. Pigment changes can be either hypo- or hyperpigmentation. Telangiectasia is frequent in all forms of SSc, it develops primarily in the face and extremities. Often the pruritus is severe and bothersome to the patient, a variety of ointments and antihistamines can be tried but rarely help. Pruritus normally improves with time.
The phenomenon of sclerodactyly disables the patient. The fingers swell and cannot be bent. On the phalanges, the skin shrinks and seems to adhere to the bone. This irreversible disability can also be seen in the feet.
8.1.2 Gastrointestinal involvement
Next after cutaneous lesions, gastrointestinal dysfunction is the next most common feature of SSc. Dysmotility of the lower two thirds of the esophagus is present in over 80% of the patients. It results from weakness of
esophageal smooth muscle, at least half of them being clinically symptomatic with retrosternal burning (61) . Reflux of gastric contents into the distal esophagus is common (68) . Esophageal involvement can be diagnosed by cineradiography (4) . Proton pump inhibitors can decrease the effect of this problem.
Small intestine motility is very frequent in patients with SSc, 40% have some kind of abnormality (13,16). Hypomotility of the duodenum results in postprandial abdominal pain, bloating and vomiting. This is similar to the changes seen in the small intestine leading to diarrhea and weight loss. The small bowel hypomotility results in bacterial overgrowth (27,80) and interference with fat absorption, which leads to intermittent periods of abdominal distention and vomiting. Antibiotic treatment can prove helpful. The colon hypomotility leads to severe obstirpation. Muscular loss of the rectal muscle may lead to incontinence and rectal prolapsed, both very bothersome for the patient.
8.1.3 Mucoskeletal involvement
Patients often suffer from polyarthralgias and morning stiffness in both small as well as large joints (86) . SSc patients often suffer from a prominent polyartritis (inflammation of the joints) and algias (often burning and painful sensation, due to psychological and inflammatory reasons) early in their illness (11) .
The most common form of myopathy (muscular affections) can be seen in 80% of the cases, and is characterized by an indolent chronic myopathy which create muscle weakness. This myopathy which persist for years is normally not treatable neither with glucocorticoid nor with steroids (44) .
Late in the disease, a resorption in the end of the distal phalanges of the fingers can occur as result of lack of vascular supply. Resorptions of different sorts can also in rare cases be seen in the mandible.
8.1.4 Pulmonary involvement
Involvement of the lungs is the fourth most common organ involvement seen in SSc patients (44) . Pulmonary interstitial fibrosis has though become the most important death cause since renal crisis has become a treatable problem (44) . In patients that develop serious pulmonary damages the greatest amount of damage can usually be seen in the first five years of illness.
The pulmonary involvement is clinically defined by the presence of bilateral basilar interstitial fibrosis on the radiographic image, pleuritis, pulmonary hypertension and or reduced diffusion capacity for carbonmonoxide. The two most frequent abnormalities that develop in patients with SSc are diffuse interstitial pulmonary fibrosis and pulmonary vascular disease with medial hypertrophy and intestinal proliferation in the pulmonary arterioles (3,70,79,80,84,93,99). The development of vascular or isolated lung hypertension due to lowered diffusing capacity of carbon- monoxide is the most deadly complications known in SSc (70,79,80,84,93,99) .
Treatment with potent vasodilators and lung transplants give some hope to previously totally untreatable disorders.
The most common abnormalities are characterized by restrictive defect with decreased lung volumes and depressed diffusion capacity in 50-73% of the cases (49).
8.1.5 Cardiac involvement
Pericardial disease attends are usually clinically silent. The prevalence of these pericardial involvement's has been described in the literature of clinical studies to be 5-16%. (3) At autopsy the prevalence has been found to be higher, 33-72% are the numbers proposed (5,16,67,79) . Pericardial involvement in SSc patients can be subdivided into to clinical forms. Either a slow onset of dyspnea with pericardial effusion of various degree (69), or as seen more seldomly an acute form with pleuric pain, fever and pericardial rub (54) .
Myocardial involvement is seen in 50% of autopsy material(6). The clinical symptoms are very non-specific but when it does occur, it has a bad prognostic (59).The involvement shown are non-symptomatic left ventricular dysfunction, conduction defects, arythmia. Patchy replacement of the myocardium and the conduction system by fibrous tissue occur in most patients (7,16, 23,25,43,67) .
8.1.6 Renal involvement
Until recently renal involvement constituted the most deadly complication of systemic sclerosis (59) . But with toady's use of angiotensin converting enzyme (ACE) inhibitors, it is now a treatable malfunction (92,95) . Patients that within 5 years of illness shows symptoms and having rapid progression are at highest risk for developing renal associated problems. Renal insufficiency is always an acute event occuring without warning.
Captopril, an ACE-inhibitor, is the drug of first hand choice (82,92) .
8.1.7 Other organs
Other common features of the manifestations of SSc are dry eyes and dry mouth. Parotidis gland enlargement and Sjögren`s syndrome will be further discussed in Chapter 9.2.2 Male impotence and libido are also reported. (17)
8.2 Histopathology
The histopathology does not differ SSc from localized scleroderma. The difference is drawn by the involvement of internal organs. However, in early lesions of SSc the inflammatory reaction is less pronounced, only a mild inflammatory infiltrate is present. But in contrast in the late stage, SSc shows more vascular changes, particularly in the subcutis. These changes are paucity of blood vessels, thickening of their walls, and narrowing of the lumen. Aggregates of calcium may also be present within the sclerotic collagen of the subcutis tissue (81)
8.3 Treatment
Although numerous pharmacological agents have been proposed in tests of treatment of SSc, no one has clearly showed good results. D'Angelo, lists these different pharmaceutical drugs which have approved effective in different studies, no major changes in therapy has been suggested since then (Tab.4) (16) .
Several vasoactive agents have been used, especially in patients with LSSc and Raynaud's phenomenon. Immunosuppressive agents have been tried out as well. No cure exist at present but effective treatment eases the symptoms. The acute myositis is systemic sclerosis patients are usually responsive to glucocorticoid treatment, but once the chronic indolent myopathy is present it has no longer an effect. Although a vast amount of drugs have been suggested there is still no drug available which alters the course of the illness.
 Table 4. Pharmaceutical drugs tested in SSc. Adapted from D'Angelo (16) .
Table 4. Pharmaceutical drugs tested in SSc. Adapted from D'Angelo (16) .
9. Orofacial manifestations of systemic sclerosis
9.1 Introduction
As described in previous chapters, SSc is a well-known disease to general medical doctors and to internal medicine specialists. Its oral and facial manifestations are less known. That is also the reason why rather little has been written and published dealing with the oral manifestations. A second reason for this may also be that these manifestations occur rather late in the process of the disease, and next to the problems caused to the patient's general condition the oral manifestations are not life threatening. Naturally the rarity of the SSc also hampers further research. The orofacial manifestations are often very specific and are often characterized by osteolysis of the mandibular ramus. This osteolysis was first described by Taveras in 1959, and has been reported in several publications after (19,31,62,63,64,65,71,98) . Even though the skeletal resorbtion is the most established stomatological pathology seen in SSc patients, other affections seem to be more bothersome to the patient (2,31,101) . The most symptomatic problems seems to be such as xerostomia or microstomia. Many patients have in interviews said to have been refused dental care due to the limited moth opening abilities.
The following chapters will discuss documented pathologies in the maxillofacial area in patients with SSc, their effects on the patient and appropriate management. All articles that are reviewed in this chapter has based their research on patients who fulfilled the diagnostic criteria's for scleroderma (87) according to the subcommittee for scleroderma.
9.2 Clinical manifestations
This chapter will present the oral manifestations in the following order:
-
Microstomia
-
Salivary glands, Sjögren`s syndrome and xerostomia,
-
Periodontal diseases
-
Dental disease
-
Mucosal and skin disease
-
Osseous resorption
9.2.1 Microstomia
Limited mouth opening or microstomia is a well documented effect of the sclerosis of the facial skin in patients with SSc. As described in chapter 8.1.2, this skin sclerosis leads to a mummified facial skin with an expressionless face. The elasticity of the facial skin gets replaced by a smooth leather appearance with restricted mouth opening (19,63,82) .
This limitation is seen in between 68-100% of the diagnosed SSc patients. This change in perioral tissue often reduces the range of motion of the mandible and hamper intubation, mastication, speech and oral hygiene and, as mentioned above, dental treatment.
This change in oral aperture of course has plenty of physic social effects. The difficulties in keeping a good oral hygiene and the following difficulty to undergo necessary dental treatment has dramatic effects. Several studies have been published about this specific symptom and suggestions of both surgical and non surgical treatments have been made. Despite this, the problem remains one of the most difficult ones in oral management of SSc patients.
Mouth opening capacity is measured in the following ways, according to Wood et al (98) :
-
. Intercommissural distance. It's measured with the teeth in occlusion and registration of the distance in between a point a along the vermilion border of the lower lip from one commissure to the other.
-
. Maximum oral aperture. Measuring the distance between the lower and upper lips vermilion line while the patient opens up as widely as possible.
-
. Interincisal distance. With the teeth in maximal intercuspation, a mark is made on the lower incisors to register the overbite, then the mouth is opened maximally and the distance between the mark and the upper incisal edge is measured.
Maximal oral aperture may range in healthy subjects, from 36 to 77mm with the mean value within 50 to 60 mm normal range.
Surgical treatments has been proposed and tried with various results. Surgically the method is relatively simple. The idea is to detach the fibrotic fibers and regain a certain elasticity. Long term the procedure has to be redone because of the ongoing fibrosis of the tissue (47) .
Nayor et al (66) published positive results from a non surgical treatment study. The method was simple and included mouth stretching and gymnastics with fingers and a tongue depressor. All of the patients in the study improved their mouth opening abilities. This can be seen as an easy non aggressive treatment and should be first hand choice in the treatment of microstomia. Drugs such as non steroid inflammatory substances can be useful in providing symptomatic pain control.
9.2.2 Salivary glands, Sjögren's syndrome and xerostomia
Mouth dryness is subjectively one of the most troublesome attaints in the facial region of SSc. Oral xerostomia is frequently found in patients with SSc, it's found in about 70% of the SSc subjects versus 33% of the control group (98). Decreased salivary flow can create problems for patients who are completely or partially edentulous. The ability to wear removable prosthesis may be compromised because of xerostomia. There is also an increased difficulty in fabricating the removable prosthesis due to both the xerostomia and microstomia.
Candida albicans can also become an opportunistic organism with or without the presence of a prosthesis (63) . Oral candidosis can be long and difficult to handle.
Sjögren's syndrome is an autoimmune disease of the exocrine glands that tends to primarily involve the salivary and lacrymal glands. Therefore the most common manifestations are dry eyes, dry mouth and glandular swelling (21,62) . Sjögren's syndrome occurs in a primary and secondary forms. Primary Sjögren's syndrome is diagnosed when a collagen-vascular disease is absent, and is characterized by a nodular lymphocyte infiltrate
the synthesis of IgM is normally considered as a sign of the Sjögren's syndrome (37) . Secondary Sjögren's syndrome occurs in patients with systemic sclerosis and also rheumatoid arthritis, lupus erythematosus and inflammatory myopathy. Salivary gland hypofunction occurred in 14 of 32 patients with SSc studied by Nagy et al (62) and frequently correlated with keratoconjnctivitis sicca. The parotid gland is often seen to be swollen in systemic sclerosis patients. Treatment for xerostomia includes artificial saliva, sugar free chewing gums and perfect oral hygiene. Principally the idea is to either increase the decreased salivary flow or to constitute saliva by artificial fluids. Substances as plaquenil and hydroxychloroquine sulfate is said to have a positive effect (63) .Dryness and cracking of the lips are treated with regular applications of petoleum-based compounds. Drugs giving additional xerostomia effect should be avoided such as anti- histamines, antispasmodics, and tricycle antidepressants. Patients should carry water with them and avoid dry foods, alcohol and tobacco. Oral corticosteroids are not believed to have any effect and should therefore be avoided (33) . Pilocarpine HC1 is a parasympathomimetic drug that functions primarily as a muscarinic-cholinergic agonist with mild beta-adrenergic stimulatory properties. Pilocarpine increases salivary output and is effective in relieving oral dryness.
9.2.3 Periodontal disease
The above mentioned microstomia and xerostomia often result in an elevated plaque index. This elevated plaque index of course resulting in gingival inflammation, gingivitis. Accordingly gingival and even mobility indexes has shown significantly higher in patients with SSc (98) . It is believed that the decreased vascularity and tissue ischemia may cause the increased periodontal disease and following tooth mobility. Patients often show signs of gingival recession and stripping of the buccal attached gingival tissues (51,63) . Even the width of the periodontal ligament is significantly increased in the SSc patients. This is also a possible factor behind the increased tooth mobility. A correlation in between the width of the periodontal ligaments and the number of organs involved by the fibrosis has been described, but neither age of the patient nor duration of the disease seem to have an effect on the width of the ligaments. Posterior teeth though showing more involvement than anterior teeth (3,8,10,19,62) . Subsequently the severity of the disease seems to be determine the periodontal involvement.
Primary treatment of the periodontal disease of patients with SSc does not differ from normal periodontal therapy. This suggesting improving plaque control with intradental cleaning as first step. Professional cleaning of supra-and-subgingival root surfaces and hereby eliminating the bleeding on probing, which is the clinical sign of an on going periodontal illness. Best results are naturally obtained with prophylactic measures.
9.2.4 Increased frequency of decayed missing and filled teeth
The limited mouth aperture due to the fibrosis of the facial skin leads to increased amount of dental plaque in the oral cavity specially around the teeth and on the back of the tongue.
Human saliva has many functions of which the elimination of food rests and dental plaque is one. Saliva's buffer capacities, that is increasing the oral pH "buffering" and therefore decreasing the acidity after food intake is another. The dental plaque older than 48 hours changes its microflora from aerobe to mainly anaerobe bacteria's. These bacteria's of which Streptococcus mutans play an important role can ferment even in low pH environment, thus creating an all time low pH. The dental crown enamel starts to dissolve itself in pH lower than 5,2 and the dental root surface has its critical pH already at 6,7 pH.
The tongue which often shows diminished mobility due to fibrotic changes loses its ability of doing mechanical cleansing of dental plaque. Due to dysphagia proper nutrition can be impossible to achieve. In conjunction with xerostomia its an initiating point of the degenerative process with increased caries and gingival problem. The xerostomia, the microstomia and the tongue immobility leads to drastically higher caries risk. This increased caries risk subsequently leads to more filled teeth surfaces. The limited mouth aperture limits the possibility to get appropriate conservatory dental care, leading frequently to drastically terapheutic measure such as tooth extraction instead of conservation. Further on the problem persists with the difficulties in replacing missing teeth even with normally simple removable prosthetics.
Primary treatment is conventional caries prevention. Perfect dental hygiene surveyed by professionals. An increase in salivation (chap 9.2.2) Local Fluor treatment in office and at home with custom made fluoration trays can reduce the caries risk, these should be fabricated already in the initial treatment plan face. Dietetic advises excluding too frequent food intakes, sugar and restriction in acid food intake. Restorations with glass ionomer with cariostatic properties can be useful. The use of chlorhexidine gluconate rinses can be useful for patients with gingival inflammation. Motivation remains the most important tool in the treatment.
Dental implants have been tried out as replacements for missing teeth, this treatment is also very limited due to the microstomia and the changes in the bone quality and quantity (63).
9.2.5 Mucosal and skin changes
Oral candidosis is a frequent opportunistic microorganism in patients with SSc. It is believed to be due to the esophageal dysmotility (63) , but it is more probable favored to xerostomia and naturally by removable prosthesis. 30-40% of the SSc patients shows microstomia with changes in the perioral skin (19). The changes seen are constricted tissue giving labial rhagades. Typical face characteristics are sclerotic skin in the palpebral, chin and upper lip region, and a thin fine nose. Telangiectasia is often up to 56% (62) seen on both lips and on the tongue. A notable decrease in tongue size with an accompanying limitation of speech and deglutition is seen in 25% of the patients (65) often giving a retrognate profile. When involved the oral mucosa often becomes rigid, thin, and may be tender to palpation.
Treatment of oral candidosis infection can be rather challenging, regardless of the usage of removable prosthesis or not. The traditional treatment include an improved hygiene, in case of removable prosthesis patient should be advised to sleep without, and use of antifungeal agents such as nystatin, mycostatin etc. Always take a microbiological sample before and after treatment to determine sensitivity and result of treatment (65) .
9.2.6 Osseous resorbtion of the mandible
According to E Wood (98), 29% of subjects with SSc shows mandibular resorption (erosions). Subjects in the very same research group showed greater mandibular erosion involvement with increasing amount of involved organs. Thus suggesting the osseous resorption increasing with severity of illness. In the above mentioned study the 29% presenting mandibular bone lesions they show average 3 lesions in the mandible. The posterioinferior angle of the mandible is the most affected area (64) , but other parts can also be affected, changes in condylar and mandibular angle that give the patient very special profile, (chap 10). A rare type of localized scleroderma can lead to Romberg's disease which represents an arrest of bone growth which leads to facial hemiatrophy. The cause of these osseous lesions has been ascribed to local ischemic processes, resulting from a narrowing of the arteriolar lumen and the increased pressure caused by the tissue sclerosis (5,11,71) .
10. Presentation of cases
The patients presented in this chapter have been seen and documented at the Division of Stomatology and Oral surgery (Professor J. Samson), at the Dental school, University of Geneva, Switzerland.
10.1 Case 1
Miss D.T was referred to our department by Professor J.H saurat at the department of dermatology at the HUG hospital for a gingival retraction. D.T had an early onset of the Raynaud`s phenomenon. She was only 12 years of age when it appeared and only 15 years old when all the fingers showed involvement. The diagnosis of systemic sclerosis was clear when she at the age of 19 started to show typical signs of SSc such as contour loss of the facial skin, a retracted mandible due to the sclerosis of the skin, incomplete lip closure, diminished mouth aperture and the typical pointy nose (Fig1,2,3) At this point a gingival retraction of the tooth 31 (Fig 4) with corresponding enlarged ligament space appeared. Radiological there was no signs of osseous resorption at this time (Fig 5).
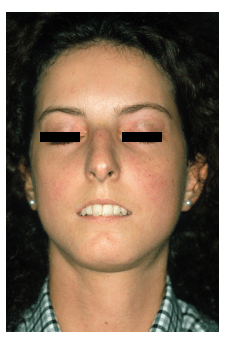 Fig 1. Female 20 years of age. Early stage scleroderma shows facial skin involvement resulting in a total change of the facial expression due to loss of the contours of the skin.
Fig 1. Female 20 years of age. Early stage scleroderma shows facial skin involvement resulting in a total change of the facial expression due to loss of the contours of the skin.
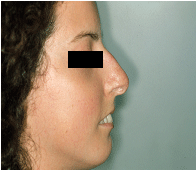 Fig 2. Incomplete lip closure acquired retrognate profile and a characteristic pointy nose.
Fig 2. Incomplete lip closure acquired retrognate profile and a characteristic pointy nose.
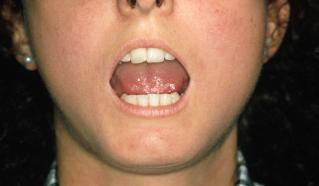 Fig 3. Limited mouth opening and decreased tongue mobility.
Fig 3. Limited mouth opening and decreased tongue mobility.
 Fig 4. Tooth 31 shows periodontal damage with corresponding gingival retraction.
Fig 4. Tooth 31 shows periodontal damage with corresponding gingival retraction.
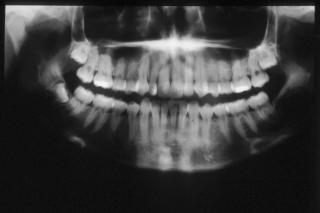 Fig 5. In this early stage of illness there is not yet a visible bone resorption other than in the alveolar bone.
Fig 5. In this early stage of illness there is not yet a visible bone resorption other than in the alveolar bone.
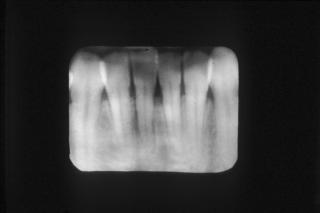 Fig 6. This shows the bone destruction and the enlarged ligament space, corresponding to the gingival retraction in Fig 4.
Fig 6. This shows the bone destruction and the enlarged ligament space, corresponding to the gingival retraction in Fig 4.
10.2 Case 2
Mrs J.A developed a systemic sclerosis at the age of 28 right after her first pregnancy. The onset was fast and aggressive in its form. The sclerosis of her hands and the claw like shape (Fig 9) it led to motorically disabled the patient's normal function enabling her to keep an adequate oral hygiene. This handicap and the decrease in mouth aperture (Fig 7,8) together with an intense hyposalivation resulted in fast developing multiple caries and periodontal damages; the flour treatment had little effect (Fig 9,10). Six years later an osseolytic process was detected in both the coronoid and condyles of the mandible. This bone loss gives the typical profile as seen in Fig 8 with a loss of contour and a retrognate intermaxillaire relation. This led to premature dental contacts of the posterior teeth that leaves the patients with an open frontal bite and incomplete lip closure. Taken together these factors made it impossible to maintain an acceptable oral function and even adequate treatment did not have any positive results. The fast progression of the systemic sclerosis led to the death of Mrs J.A at the age of 39.
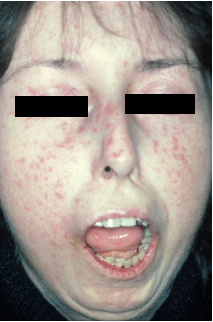 Fig 7. Female 34 years of age. Aggressive form of scleroderma with widespread skin involvement. Total elimination of the facial expression, numerous telengiectasia in plaque form, pointy nose, microstomia with a deviation due to TMJ resorption.
Fig 7. Female 34 years of age. Aggressive form of scleroderma with widespread skin involvement. Total elimination of the facial expression, numerous telengiectasia in plaque form, pointy nose, microstomia with a deviation due to TMJ resorption.
 Fig 8. Incomplete lip closure with result in upper incisive protrusion. Retrognatism, angular bone resorption evident in clinical examination
Fig 8. Incomplete lip closure with result in upper incisive protrusion. Retrognatism, angular bone resorption evident in clinical examination
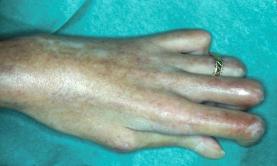 Fig 9. Claw like form of her hands.
Fig 9. Claw like form of her hands.

 Fig 10. and 11. Resorption of the condyles and mandibular ramus bilaterally, and multiple decay's. Premature contacts of posterior teeth, leading to an open frontal bite and incomplete lip closure. Note the rapid evolution showed in only one year.
Fig 10. and 11. Resorption of the condyles and mandibular ramus bilaterally, and multiple decay's. Premature contacts of posterior teeth, leading to an open frontal bite and incomplete lip closure. Note the rapid evolution showed in only one year.
10.3 Case 3
Mrs. A.C develops systemic sclerosis at the age of 50. This form will show to have an aggressive onset involving the extremities, the face and the esophagus. The facial symptoms are sclerosis of the facial skin, lip retraction, pointy nose resulting in a maxillary proalveolia and mandibulare retrognati. A severe form of hyposialia and the hypofunction of the hands enhanced the fast development of oral diseases such as multiple caries, periodontal diseases and recurring oral candidosis. Within 8 years of rapid development a clear osteolysis was seen in radiographs in the postero-inferio angels and at the metal foramen of the mandible.
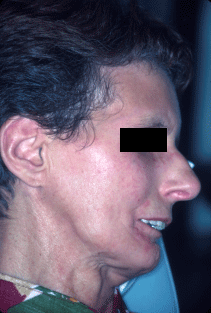 Fig 12. Pronounced retrognat profile and typical shiny facial skin. Loss of mandibular ramus shows clinically. The eye gives a sunk in expression.
Fig 12. Pronounced retrognat profile and typical shiny facial skin. Loss of mandibular ramus shows clinically. The eye gives a sunk in expression.
 Fig 13. Advanced angular resorption bilaterally and multiple dental pathology.
Fig 13. Advanced angular resorption bilaterally and multiple dental pathology.
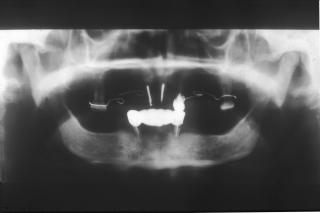 Fig 14. Shows rapid development of the angular and ramus resorption within an 8 year period.
Fig 14. Shows rapid development of the angular and ramus resorption within an 8 year period.
10.4 Case 4
Miss C.A developed at the age of 10,5 years a localized form of scleroderma. This localized form involved the tip of the nose, the upper lip, and the gingival mucous, and the alveolar bone (Fig 15,16) Clinically this was seen as an invagination of the nose tip, the upper lip, a gingival retraction and an ankylosis of the corresponding tooth 21 with a hinder of full growth of the alveolar bone structure.
 Fig 15. Localized form of scleroderma. Here seen on the nose in a 'coup de sabre'. Involving nasal tissue the upper lip and adjacent tissue.
Fig 15. Localized form of scleroderma. Here seen on the nose in a 'coup de sabre'. Involving nasal tissue the upper lip and adjacent tissue.
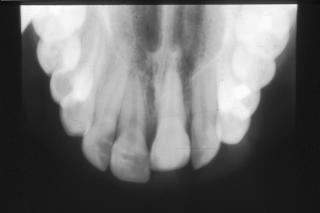 Fig 16. Ankylosis of 21, loss of ligament space.
Fig 16. Ankylosis of 21, loss of ligament space.
11. Conclusions
Systemic sclerosis is a connective tissue disease known to us for almost 200 years. The aim of this literature study was primary to create a catalogue listing all of the reported stomatological manifestations. Secondly to find out weather any local dental treatments could be suggested. From this literature study it is obvious that little progress has been done in solving the mysteries of systemic sclerosis during the last decades. The etiology is unknown but exposure to certain agents may be associated with its occurrence. Since Bramwell reported an association between scleroderma and occupations associated with silica dust in 1914 several reports have documented an unusually high prevalence of SSc in these patients, but if this is real SSc or only a scleroderma like diseases is still to be discussed. The main diagnostic tool is clinical and can easily be confused by differential diagnostics as for instance when the bilateral Raynaud`s syndrome is the only symptom and the following manifestation for several years. When the skin sclerosis is discrete it can correspond to several connective tissue diseases. Proposed differential diagnosis are angiomatosis of Rendu-Osler, which in contradiction to SSc has hereditary predilection. Transplanted immune suppressed patients can also show similar symptoms. Lichen planus is a connective tissue disease often mistaken for SSc. It is the rarity of the disease and the different degrees of involvement that hampers the studies.
There are several alterations of the skin and gums of which telangiectasia is one. Telangiectasia is not restricted to the limited form of SSc. A high number of SSc patients show a decreased lachrymal and salivary excretion rate characteristic to Sjögren`s syndrome. These decreased salivary rate is due to either fibrotic processes or to Sjögren`s syndrome.
Furthermore an increase in need of medical and dental attention is obvious in these patients. As shown in the text many of these patients suffer from a wide range of facial, oral and dental disorders. The presentation of cases should give a clinical profile of these patients and the text suggests that prophylactic treatment is the most efficient one.
[Previous]
[Next]