1. The antiphospholipid syndrome
1.1. Introduction
The antiphospholipid syndrome (APS) was first described in 1983 (Hughes). APS is an autoimmune disorder characterized by: 1) the presence of antiphospholipid antibodies (APLA): anticardiolipin antibodies (ACA) and/ or lupus anticoagulant antibodies (LA), and 2) clinical complications such as a) venous and/ or arterial thrombosis, and/or b) pregnancy morbidity (International Consensus Statement for Definite Antiphospholipid Syndrome). The syndrome is called primary APS when it occurs without underlying disorder and secondary APS in patients with other diseases such as auto-immune diseases, particularly systemic lupus erythematosus, or malignant diseases. Endothelial cell (EC) activation by APLA seems to be one of the major pathogenic mechanisms of APS. The mechanisms underlying this activation are one of the research topics of the Division of Angiology and Hemostasis. It was observed that a monoclonal antibody to late endosomes recognized the anionic phospholipid lysobisphosphatidic acid (Kobayashi et al, 1998; Galve-de Rochemonteix et al, 2000). Further investigations showed that lysobisphosphatidic acid is an important target for APLA and that APLA accumulate in late endosomes of human umbilical vein endothelial cells (HUVEC) leading to a redistribution of the insulin-like growth factor 2/ mannose-6-phosphate receptor (CI-M6PR) from the Golgi apparatus to late endosomes. It was also found that beta2 glycoprotein I (b2GPI) accumulate at the cell surface and in late endosomes in HUVEC, resulting in modification of intracellular protein trafficking as shown by a redistribution of the CI-M6PR from the Golgi apparatus to late endosomes (Dunoyer-Geindre et al, 2001). The mechanisms by which APLA induce a thrombotic phenotype were further studied in the Division. It was found that incubation of EC with anti- b2GPI antibodies resulted in a redistribution of the transcription nuclear factor kB (NFkB), an essential intermediate in the activation of EC, from the cytoplasm to the nucleus after a delay of several hours (Dunoyer-Geindre et al, 2002). This NFkB redistribution was accompanied by an increased expression of tissue factor (TF) and the leukocyte adhesion molecules ICAM-1, VCAM-1 and E-selectin (Dunoyer-Geindre et al, 2002).
Other recent data reported that EC activation by APLA lead to a proinflammatory endothelial phenotype with a monocyte adhesion to the endothelium and penetration into the subendothelial space (Pierangeli et al, 2000). Another study showed that upregulation of adhesion molecules by some murine monoclonal anti- b2GPI antibodies correlated with foetal resorption in mice (George et al, 1998).
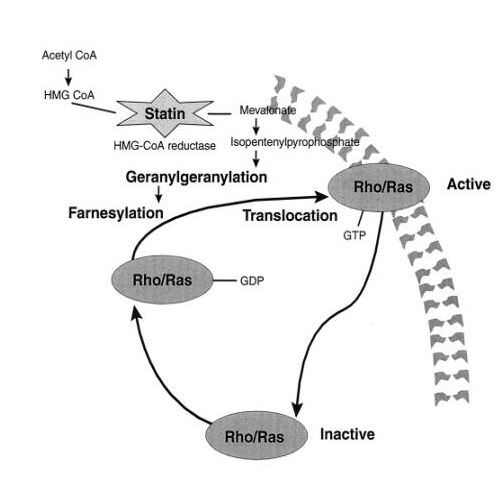
The aim of this study was to investigate the ability of statins, specific inhibitors of cholesterol biosynthesis with clinically proven beneficial effects such as regression of atherosclerotic lesions and reduction of cardiovascular complications (Scandinavian Study group, 1994; Hebert, 1997), to modify adhesion molecule expression in APLA-activated EC and to study the underlying mechanisms.
1.2. Antigens and antibodies in the antiphospholipid syndrome
1.2.1. Antigens
Recent studies demonstrated that APLA recognize not only determinants with anionic PL but also neutral PL as well as proteins (Table 1).
A) Phospholipids (PL)
Anionic PL are essential cofactors for the blood coagulation system. APLA are supposed to bind the phosphodiester group of negatively charged PL. The length and composition of the polar head groups as well the saturation of the fatty acid side chains contribute to the antigenicity (Levy et al, 1990). Possible antigenic targets of APLA on EC include negatively charged PL such as cardiolipin, phosphatidylserine, phosphatidylinositol, phosphatidylethanolamine, phosphatidic acid.
-
Cardiolipin (CL)
CL has been most often used to study the interaction of APLA with anionic PL. The antibodies that directly bind to CL are characterized by the presence of a larger than average density of positively charged arginine residues in one of antigen binding loops, which mediate the binding to the negatively charged PL. -
Phosphatidylserine (PS), phosphatidylinositol (PI)
PS and PI may be possible antigenic targets of APLA on EC. PS is a PL occurring in abundance in the inner leaflet of the cytoplasmic membrane and is exposed to the outer surface after cell activation or apoptosis (Mevorach et al, 1998). PS promotes the interaction of coagulation factors. PI derivates are important mediators of cell activation. -
Phosphatidylethanolamine (PE), phosphatidylcholine (PC), phosphatidic acid (PA)
Zwitterionic antigens such PE and PA have also been implicated in APS (Berard et al, 1996). PL have a positively charged substituted group that interacts electronically with the phosphodiester, thus blocking APLA access to the phosphodiester. APLA do not usually bind the zwitterionic molecule PC, but removal of the positively charged choline group results in the formation of negatively charged PA, which could be bound by APLA. -
Other PL antigens
Other PL can also be involved. Recently, lysobisphosphatidic acid, a lipid related structurally to CL and present in the membranes of late endosomes, has been identified as another immunologic target for APLA. The team of Division of Angiology and Hemostasis observed that APLA could react with lysobisphosphatidic acid of internal membranes of late endosomes, resulting in accumulation of APLA in late endosomes and subsequent EC activation (Kobayashi et al, 1998; Galve-de Rochemonteix et al, 2000, Dunoyer-Geindre et al, 2001). The oxidation of PL may be necessary for epitopes recognition by some APLA. Horkko et al (1997) demonstrated that many APLA bound to CL only after it had been oxidized, but not to a reduced CL analogue that could undergo oxidation. They suggested that the reactive groups of oxidized CL, such as aldehydes, generated during the decomposition of oxidized polyunsaturated fatty acids, form covalent adducts with b2GPI (and other proteins) and that they are epitopes for APLA.
Whether this broad response of APLA indicates polyclonality or crossreactivity could not be determined using sera as a source of antibody. It has been shown that a human monoclonal IgM LA is able to bind to PA, PI and PS. This provided evidence for the polyspecificity of at least some APLA (Thiagarajan et al, 1980).
Binding of APLA to PL is also influenced by the physical state of the PL. Mice immunized with hexagonal versus lamellar phase PL are more likely to develop APLA (Rauch et al, 1990). Protein cofactors like b2GPI may shift the orientation of some PL from the lamellar to the hexagonal phase.
B) Proteins
Beta2 glycoprotein I (b2GPI)
b2GPI, also called apolipoprotein H, is a major protein constituent of human plasma, where its concentration is about 200 mg/L. It is the main factor for the recognition of anionic PL by APLA.
- Structure
b2GPI is a single-chain, 5 kDa protein consisting of 326 amino acids. It contains large numbers of Pro and Cys residues, and is highly glycosylated (Figure 1). The protein is a member of the complement control protein or short consensus repeat (SCR) superfamily, characterized by repeating stretches of about 60 amino acid residues, 'Sushi domains', each with a set of 16 conserved residues and 2 fully conserved disulfide bonds. It has five repeating SCR domains. The first four homologous repeat regions consist of about 60 amino acids with 2 disulfide bonds in each domain. The fifth domain contains 80 amino acids and 3 disulfide bonds. The protein binds to phospholipid membranes via the cationic portion of its fifth SCR domain. b2GPI has a weak affinity for anionic PL but APLA, binding to domains I and V, increase this affinity, due to the bivalency of the antibodies and to conformational changes induced in the b2GPI protein.

|
(A) Ribbon drawing of b2GPI with consecutive domains labelled I-V. N-linked glycans, as well as the position of the putative O-linked glycan, Thr130, are indicated by a ball-and-stick model. The strands are shown in red and helices in green (B) Topology diagram of b2GPI. The central sheets of all five domains are labelled B2(-B2)-B3-B4(-B5), the N- and C-terminal -sheets are labelled B1'-B2' and B4'-B5', the -helix and the 3/10 helix are denoted A1 and A2 and numbers of residues delimiting secondary structure elements are given. Disulfide bonds are indicated with dashed lines. The positions of N-glycosylation are given by hexagons; a diamond indicates the putative O-glycan. Horizontal dashed lines mark domain boundaries (C) Ribbon representation of domain III of b2GPI with labelled secondary structure elements. The two fully conserved disulfide bonds are shown in yellow (D) Ribbon representation of domain V of b2GPI with labelled secondary structure elements. The three disulfide bonds are indicated with yellow lines. The aberrant face, which contains the membrane-binding site, is located on the right-hand side. |
- Functions
It was found that b2GPI interacts specifically with lipoprotein(a) and the endothelial cell protein annexin II (Ma et al, 2000). Although the physiologic importance of b2GPI anticoagulant activity is still unclear, b2GPI acts at least in vitro as an inhibitor of the intrinsic blood coagulation pathway due to its ability to interact with negatively charged surfaces, which in turn are necessary for the activation of factor XII (Schousboe et al, 1985). Interestingly Mori et al (1996) have shown that b2GPI can inhibit the anticoagulant activity of activated protein C. Thus, it remains unclear whether b2GPI in vivo has anticoagulant or procoagulant properties. b2GPI has been reported to inhibit adenosine diphosphate-mediated platelet aggregation and the prothrombinase activity of activated platelets (Shi et al, 1993). b2GPI binds to cells undergoing apoptosis and may be involved in the rapid, noninflammatory clearance of these cells by phagocytes. Recent study showed that the opsonization of apoptotic cells with anti-b2GPI antibodies may be a proinflammatory event, stimulating the presentation of apoptotic cell antigens by dendritic cells. Thus, anti-b2GPI antibodies might possibly contribute to an autoimmune response (Rovere et al, 1999). In addition, b2GPI binds to the cell surfaces by binding to negatively charged molecules such as anionic PL, heparan sulfate proteoglycans and preferentially to oxidized low density lipoproteins. This property has been proposed to be clinically relevant providing a link between anti-b2GPI antibodies and atherogenesis (Matsuura et al, 1998).
Vitamin K-dependent proteins
-
Prothrombin (Pt)
Pt is a vitamin K-dependent glycoprotein with a molecular weight of 72 kDa. It is activated to thrombin by the so-called "prothrombinase complex" (coagulation factors Xa, Va, and calcium on a procoagulant PL surface) by the cleavage of two or possibly three peptide bonds. The first proteolytic cleavage leads to fragment 1,2 and prethrombin 2, which is further proteolysed to thrombin. The anti-Pt antibodies may react both with Pt and its fragment 1,2, but not with the decarboxylated molecule. Antibodies to Pt are found in 50-90% of patients with LA (Galli et al, 1997; Kandiah et al, 1998) but their clinical relevance is still controversial (Galli et al, 1998). Anti-Pt antibodies may inhibit the activation of Pt into thrombin. They are therefore expected to be anticoagulant. However, Vaarala et al (1996) found that anti-Pt antibodies were associated with a hypercoagulable state and high levels of anti-Pt antibodies predicted a 2.5-fold increase in the risk of myocardial infarction or cardiac death (n =106). In contrast, other investigators (Horbach et al, 1996) reported that the presence of anti-Pt antibodies in LA positive patients (n = 60) does not increase the risk for thrombosis. -
Protein C, protein S, thrombomodulin (TM)
These proteins are components of the protein C anticoagulant pathway. In this pathway, thrombin binds to TM and loses its ability to convert fibrinogen into fibrin. Furthermore, it acquires the ability to activate protein C which, in the presence of PL and protein S, degrades the coagulation factors Va and VIIIa and blocks further thrombin generation. Deficiency of protein C and S increases the risk of venous thrombosis (Oosting et al, 1993). Pengo et al (1996) investigated 22 patients with IgG APLA and thrombosis and found elevated levels of anti-protein C IgG and anti-protein S IgG in 18% and 55% of the patients, respectively. Antibodies to TM were found in 30% of the patients with a LA (n = 58) and 10% of the patients (n = 200) with unexplained thrombosis (Carson et al, 2000).
Annexins
-
Annexin V (human placental anticoagulant protein 1)
Annexin V is an anionic PL-binding protein, expressed by placental and vascular endothelium. It is postulated that annexin V plays a thromboregulatory role at the vascular-blood interface by shielding anionic PL from forming a complex with coagulation proteins in the circulation. Annexin V could play an important role in the clinical manifestations of APS, particularly in obstetrical complications (Rand et al, 1999; Rand et al, 2002). -
Annexin II
Annexin II has not been presently implicated as a target antigen of APLA. However, it was identified as a high affinity b2GPI-binding protein at the surface of EC, another mechanism of b2GPI endothelial cell binding being through the putative PL-binding site located in the fifth domain of the molecule (Meroni et al, 2001). Annexin II does not span the cell membrane, its involvement probably requires an unknown adapter protein (Ma et al, 2000).
Other proteins
Other potential protein targets for APLA include various proteins such as factor X, high molecular weight kininogen, factor XI, and the protein core of heparan sulfate (Shibata et al, 1994).
In summary, different antigens have been identified in APS. It is now known that antigen targets are mainly proteins such as b2 GPI, Pt or TM forming eventually a complex with various phospholipids.
1.2.2. Antibodies
APLA comprise a broad family of autoantibodies that includes both LA, detected by coagulation tests, and ACA, detected by ELISA methods (Pierangeli et al, 2001).
A) Immunoglobulin classes
Previous data have shown that the strongest associations of ACA and anti- b2GPI antibodies with clinical manifestations of APS involve antibodies of the IgG isotype. Cohen et al (1993) determined in vivo that immunisation of mice with pathogenic IgG and IgM ACA, isolated from the serum of a patient with APS, induce the production by the mice of anti-ACA with ACA activity. The mice developed overt APS. IgG ACA were found to have higher pathogenic potential than IgM ACA. However, several works suggest that IgM as well as IgA may also be associated with the disease, although to a lesser extent. Anti-b2GPI antibodies of IgG, A, and M classes have been reported in 84.8%, 59.3% and 51.5% of patients with primary APS, respectively (Lacos et al, 1999). Amoroso et al (2003) evaluated the prevalence of IgG and IgM antibodies to various antigens in sera from 87 patients affected by SLE. IgG ACA, IgG anti-PA, IgG anti-PI, IgG anti-PS, and IgG anti- b2GPI were found in 53%, 37%, 32%, 38%, and 24% of patients, respectively. IgM-ACA, IgM anti-PA, IgM anti-PI, IgM anti-PS, and IgM anti- b2GPI were detected in 15%, 17%, 18%, 14%, and 16%, respectively.
-
IgG
The association of thrombotic risk with high-titer IgG ACA or anti- b2GPI antibodies, often together with LA, has been confirmed in several studies (Gattorno et al, 1995; Silver et al, 1996). Sammaritano et al (1997) investigated whether the presence of ACA of a specific IgG subclass is associated with clinical complications of APS. They found that IgG2 was the predominant subclass of ACA, detected in 75% of the patients, and it was significantly associated with thrombotic complications. The IgG subclass distribution of anti- b2GPI and ACA in patients with primary APS and secondary APS was studied recently (Samarkos et al, 2001). Mean values for anti- b2GPI antibodies were as follows: IgG1- 24.4%, IgG2- 70.2%, IgG3- 5.0%, IgG4- 0.4%. The reported IgG subclass distribution for ACA was: IgG1- 40.1%, IgG2- 32.8%, IgG3- 23.7%, IgG4- 3.3%. Comparing the ranking of IgG subclasses of anti- b2GPI antibodies (IgG2 >IgG1 >IgG3 >IgG4 ) and ACA (IgG1 >IgG2 >IgG3 >IgG4 ), IgG2 was the most prevalent subclass for anti- b2GPI antibodies whereas for anti-CL antibodies IgG1, IgG2 and IgG3 were all frequently elevated. An association between IgG2 and IgG3 anti-b2GPI antibodies and venous thrombosis has been shown, while IgG2 and IgG3 ACA were found to be more specifically associated with arterial thrombosis. -
IgM
In a study determining the distribution of ACA in primary and secondary APS, IgM were found in 26% of secondary APS cases and in 15.8% of primary APS group (Vianna et al, 1994). The antibodies of IgM isotype were related mainly to thrombocytopenia and heart valve disease (Diri et al, 1999). APLA, associated with infections, typically are IgM isotypes and are usually transient (Jaeger et al, 1992). -
IgA
Evidence that IgA antibodies may be important continues to accumulate (Lacos et al, 1999). A significant relationship has been demonstrated between increased IgA levels and a history of venous thrombosis, thrombocytopenia, heart valve disease, livedo reticularis, and epilepsy. Interestingly, black patients with primary and secondary APS appear to have a higher frequency of IgA anti-b2GPI antibodies, suggesting genetic predisposition for these antibodies (Diri et al, 1999). -
Coexistence of different classes of APLA in the same patient
It has been recently reported that the concurrent presence of IgG, IgM, and IgA appears to increase the frequency of recurrent spontaneous abortions as compared with the presence of a single isotype among the autoantibodies (Guglielmone et al, 1999). Vogel et al (1991) determined APLA in an unselected group of 63 SLE patients. They found APLA in 50.8% of the patients and the simultaneous presence of LA and ACA was associated with an increased of arterial thromboembolic events.
In summary, various studies have demonstrated that APLA are a large and heterogeneous family of immunoglobulins from IgG, IgM, and IgA classes and that the simultaneous presence of APLA increases the risk of clinical complications.
1.3. Laboratory detection of apla
1.3.1. History
In 1952, Conley and Hartmann published the first description of two patients with SLE and an unique PL-dependent coagulation inhibitor characterized by prolongation of the whole blood clotting time and the prothrombin time. Both patients had a biologic false-positive serological test for syphilis with no evidence of infection. The antibodies responsible for the false-positive serological test for syphilis were ultimately found to recognize CL within the test reagent. In subsequent years, several authors described similar inhibitors that interfered with prothrombin time activation, but without specificity to any known coagulation factors; a number of these patients also had false-positive serological test for syphilis (reviewed in Shapiro et al, 1982). In 1957, Laurell and Nilsson described an association between chronic biologically false-positive serological test for syphilis, a circulating anticoagulant, and recurrent pregnancy loss. A paradoxal association between PL-dependent coagulation inhibitors and thrombosis was first recognized by Bowie et al in 1964. They described the presence of thrombotic complications in 4 of 8 SLE patients having this inhibitor. The term «lupus anticoagulant» (LA) was proposed in 1972 to describe these inhibitors on the basis of their prevalence in SLE patients (Feinstein et al, 1972). In 1980, the immunologic nature of interaction of LA with anionic PL was demonstrated by isolating an IgM paraprotein with LA activity from a patient with macroglobulinemia (Thiagarajan et al, 1980). This paraprotein inhibited the Ca2+ -dependent binding of prothrombin and factor X to PS-containing liposomes, explaining the extremely prolonged PL-dependent coagulation tests seen in the patient. This paraprotein did not interfere with the binding of factor Xa to platelets, suggesting an explanation for the fact that this patient, like most patients with LA, had no bleeding tendency. Subsequently, it was observed that most LA reacted with CL, which was used as the antigen in serologic tests for syphilis (Pengo et al, 1987). Based on these observations, in 1983 Harris et al developed a radioimmunoassay and, subsequently an ELISA for ACA demonstrating the overlapping specificity of LA, ACA and other APLA.
The complex specificity of ACA became apparent in 1990 when several groups found that actually the majority of ACA in ELISA required the presence of a plasma protein, b2GPI, in addition to anionic PL (Bevers et al, 1991; Galli et al, 1991; Koike et al, 1991). Subsequently, antibodies to a number of PL-plasma protein complexes have been described, involving among others prothrombin, protein C and annexin V, all binding to anionic PL.
Actual laboratory and clinical criteria for APS were formulated during the workshop in Sapporo, 1998, following the Eigth International Symposium on APLA.
1.3.2. Laboratory criteria for APS
Definite APS is considered to be present in a given patient when at least one of the clinical criteria (see chapter IV) and at least one of the main following laboratory criteria are met:
- ACA of IgG and/or IgM isotype in blood, present in medium or high titer, on 2 or more occasions, at least 6 weeks apart, measured by a standardised ELISA for b2GPI-dependent ACA,
-
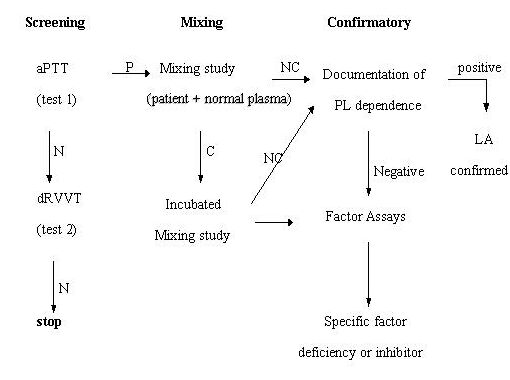
LA present in plasma on 2 or more occasions at least 6 weeks apart, detected according to the guidelines of the International Society of Thrombosis and Hemostasis (Scientific Subcommittee on Lupus Anticoagulants/ Phospholipid-Dependent Antibodies, 1995), in the following steps (Figure 2):
- Prolonged PL-dependent coagulation demonstrated on a screening tests: activated partial thromboplastin time (aPTT), kaolin clotting time (KCT), dilute Russell's viper venom time (dRVVT), dilute prothrombin time (dPT), Textarin time.
- Failure to correct the prolonged coagulation time on the screening test by mixing with normal platelet-poor plasma.
- Shortening or correction of the prolonged coagulation time on the screening test by addition of excess PL.
- Exclusion of other coagulopathies, e.g., factor VIII inhibitor or heparin, as appropriate.
|
Tests need to be repeated on 2 occasions, at least 6 weeks apart. N: normal, P: prolonged, C: correction, NC: no correction |
The sites of action of the different assays are presented on Figure 3.

|
PL: phospholipids, aPTT: PL:activated partial thromboplastin time, daPTT: diluteactivated partial thromboplastin time, KCT: kaolin clotting time, SCT: silica clotting time, DRVVT : dilute Russell's viper venom time, TTI: tissue thromboplastin inhibition. |
1.3.3. Immunologic assays for ACA
ACA react with CL and other anionic PL in solid-phase immunoassays. ACA are commonly detected by ELISA. Microplate wells are coated with CL, blocked with a solution of animal serum diluted in buffer, and then incubated with dilute patient serum. The bound antibodies onto the coated wells are then detected using enzyme-conjugated, isotype-specific secondary antibody and a chromogenic substrate (Loizou et al, 1985).
-
Isotypes
Reference sera that are isotype specific (IgG, IgM, and IgA) are now available. IgG and IgM ACA concentrations are expressed in units. By definition, one unit represents cardiolipin binding activity of 1 mg per ml of affinity-purified ACA antibody from reference sera. IgM is measured in MPL units (1 MPL unit = 1 µg of affinity-purified IgM ACA from an original reference serum) and GPL units are used for IgG (1 GPL unit = 1 µg of affinity-purified IgG ACA from an original index serum sample). -
Titer
Reporting the test results, it is important to identify the titer together with the isotype. The titer of ACA can be presented as high, moderate, low, or negative. In many cases of moderate or low titer, ACA are transient antibodies as a result of intercurrent infections. Therefore, it is important that the initial positive result is repeated after 6-8 weeks. Persistence of ACA is one of the criteria necessary to establish the diagnosis of APS. The identification of a positive test result depends upon the laboratory's care in identifying an appropriate normal reference interval. The distribution of the normal range is logarithmic rather than Gaussian. In order to quantify ACA, four house standards from two University Hospital laboratories were compared with the standards provided by the Antiphospholipid Standardization Laboratory by using two different plates and two different buffered protein solutions. Slopes from the serial dilutions of each of the four house standards were found comparable. In contrast different slopes were obtained when using the ASL standards which consist of a mixture of sera. These results indicated that dilutions of single sera are more suitable than mixture of sera when quantification of ACA is required (Rupin et al, 1994). The team from Haemostasis Unit at University Hospital of Geneva studied the positivity variation for ACA in 61 patients and 42 controls by comparing two commercial kits (A and B) with their own assay (C). The results were as follows: 50.8% ACA positivity for A, 57.4% for B and 50.8% for C. As for controls the concordance in patients was better between kit A and assay C than between kit B and assay C (de Moerloose et al, 1990). The performances of nine commercial kits and an in-house method for the quantitation of ACA have been evaluated in a multicenter study (Reber et al, 1995). Marked differences in positivity rate between kits were observed, ranging from 31 to 60% for IgG and 6 to 50% for IgM. This study showed that differences in positivity rates between the commercial kits might contribute to the differences in ACA prevalence rate found in the literature. The choice of cut-off levels might partly explain the moderate concordance between the kits. Sustained efforts on anticardiolipin standardization have resulted in improved agreement between commercial assays.
The ACA ELISA is a sensitive test, but its disadvantage is that it may be positive in a number of disorders other than APS. Alternatively, newer assays that use b2GPI or a mixture of negatively charged PL (APL ELISA Kit) have been proposed for more specific measurements of antibodies present in APS (Roubey et al, 1995). The epitope recognized most often by ACA is thought to be b2GPI. Thus, ELISA kits are using b2GPI to distinguish autoimmune ACA from true ACA that do not require b2GPI for direct binding to CL. Comparisons for the sensitivities and specificities of the standard ACA ELISA, anti-b2GPI ELISA, and APL ELISA Kit were made (Pierangeli et al, 2001). They showed consequently 100%, 74%, and 98% sensitivity of the three assays for APS. Their specificity was, as follows: 60%, 82% and 99%. Inter-laboratory variability of anti-b2GPI measurements (IgG and IgM) was investigated in the frame of the European Forum on Antiphospholipid Antibodies and its Standardization Group (Reber et al, 2002). They found that the rate of positivity varied from 50% to 93% for IgG and from 13% to 70% for IgM anti-b2GPI. Excellent concordance between centers occurred only in 13% of cases for IgG and in 6% of cases for IgM, because many selected samples were low-positive. Despite the large variability of anti-b2GPI measurements between centers, the authors found a good agreement with high- and medium-positive samples. According to an agreement reached during the eighth International Symposium on Antiphospholipid Antibodies (Wilson et al, 1999), the LA test and ACA ELISA should be used primarily in the diagnosis of APS. Moderate-to-high positive ACA or LA test results and well-documented clinical features are enough for diagnosis of APS to be made. If these test results are negative or equivocal, more specific tests, such as anti-b2GPI ELISA or anti-PL ELISA Kit might be used to confirm the diagnosis of APS.
1.3.4. Coagulation assays for LA
LA are defined as immunoglobulins that inhibit PL-dependent coagulation tests in the absence of specific coagulation factor inhibition. Generally, LA are suspected when one of several screening assays, most commonly aPTT, is prolonged. A suspected LA is further evaluated using an inhibitor screen (mixing study), in which the aPTT of a mixture of the test and normal plasma is measured. LA usually prolong the clotting time of normal plasma immediately, but on occasion prolongation requires incubation (Clyne et al, 1988). If the initial mixing study shows correction of the abnormal clotting time, then an assay for time-dependent inhibition should be performed. A diagnosis of LA should not be made on the basis of multiple abnormal screening assays and mixing studies only. If the mixing studies indicate the presence of circulating inhibitor, then LA confirmatory studies should be performed. Factor assays may be used when mixing studies show correction, suggesting a factor deficiency, when the LA confirmatory studies are negative or when a specific factor inhibitor is suspected (Brandt et al, 1995).
A) Screening assays for LA (Table 2)
The presence of LA is often first suspected when the aPTT is abnormal and fails to correct with normal plasma. Among screening coagulation tests, the aPTT is more sensitive than PT, probably because of the lower PL content of the reagent used in aPTT. A common concept is that the amount of PL in the test system is a critical determinant of sensitivity. Test systems with reduced amount of PL such as daPTT, KCT, dRVVT, dPT thus might offer the possibility of increased sensitivity due to their low PL concentration (Working Group on Haemostasis of the "Société Française De Biologie Clinique", 1993). The Textarin time was found to be the most sensitive screening test for LA when compared with the other test systems. Data show that the PL concentration is not the only determinant of assay sensitivity to LA. Altering the incubation time of aPTT, rather than the PL concentration, was a sensitive way of detecting LA (Robert et al, 1994) but it does function with few cephaloplastins. Furthermore, for partly unknown reasons, some assays appear to be more sensitive to certain subgroups of LA. Therefore, at least two different types screening assays must be performed before the presence of LA can be ruled out. The presence of platelets or platelet fragments in plasma after centrifugation can affect the results of coagulation tests, particularly when plasmas are frozen before testing, and the use of twice centrifuged plasma, or filtration through a 0.2-mm filter, has been advocated to avoid this problem (Ames et al, 1996). This is particularly important for assays without added PL (KCT) or with very low amount of PL (dPT). The residual amount should not exceed 10 G/l (Exner et al, 2000).
The protein dependence of LA may affect the tests differently. For example, it has been shown that KCT is most abnormal in the presence of Pt-dependent antibodies (Galli et al, 1995), whereas the dRVVT is mainly abnormal in the presence of b2GPI-dependent antibodies.
B) Mixing studies for LA
The presence of an inhibitor is usually documented by mixing patient plasma with normal plasma and by demonstrating a persistence of an abnormal clotting time. The sensitivity of LA testing depends on the ratio of patient plasma to normal plasma used to detect the anticoagulant effect, and these variations have not been standardized. The most common ratio of patient to normal plasma, used in mixing studies, is 1:1. However, Clyne et al (1993) showed a relatively high incidence of negative mixing studies using a 1:1 ratio in aPTT system. Some authors have suggested 4:1 mixture for evaluation of mildly prolonged aPTT (McNeil et al, 1991). Other groups have proposed a 1:4 mixture for some cases (Petri et al, 1997).
The problem with negative mixing studies may be related to the observation that some LA show time-dependent inhibition in clotting assays. Up to 15% of LA may show correction of the prolonged clotting time if tested immediately after mixing but a lack of correction if the clotting time is repeated after incubation of patient and normal plasma. These data have been challenged by Exner (2000), who demonstrated that upon buffering the mixture, the time dependence is abolished.
There is a lack of uniform criteria for the evaluation of mixing studies. One approach is to determine an index of correction. The basic formula for this index is:
Index= 100x(b-c)/a, where a= clotting time of patient plasma, b= clotting time of patient + normal plasma mixture, and c= clotting time of normal plasma.
C) Confirmatory assays for LA
A number of confirmatory assays based on the principle of adding to or altering the PL content of the test system have now been described (Table 3). They can be grouped according to the screening assay on which they are based. In the diagnosis of LA, it is important to use a confirmatory study that corresponds to the screening test which is abnormal.
The basis of these assays is to determine the effect of altering the PL content of the assay system, by adding platelets, platelet vesicles or hexagonal phase PL. The majority of test systems that are now employed to demonstrate PL dependence use increased concentrations of PL or frozen thawed platelets. The increased PL or platelet membranes 'neutralize' or 'bypass' LA. The platelet neutralisation procedure uses washed frozen and thawed platelets as a source of PL and is most commonly used in aPTT (Triplett et al, 1983). The hexagonal phase PL test is based on the finding that PE is capable of supporting coagulation test and can effectively neutralize LA activity. Comparing these 2 tests, platelet neutralisation procedure and hexagonal phase PL in systemic lupus erythematosus plasmas, Reber et al (1994) found that platelet neutralisation procedure gave a higher rate of detection than hexagonal phase PL and other tests of detection. Addition of platelets may also shorten the aPTT in the presence of anti-F VIII antibodies.
As shown in Figure 4, LA and ACA may occur independently or may coexist. LA and ACA activities may be due to the same antibody, or the activities may be physically separable (Champley, 1991). There is another family of antibodies, called reagin antibodies, which are observed in patients with APS. This group of antibodies reacts with the Venereal Disease Research Laboratory (VDRL) reagent. The VDRL reagent denotes a mixture of antigens, including CL, cholesterol and lecithin (Singh et al, 2001).

|
LA: lupus anticoagulant ACA: anticardiolipin antibodies VDRL: antibodies reacting with the Veneral Disease Research Laboratory reagent |
1.3.5. Indications for laboratory testing
Because of the high risk of thrombosis and miscarriage and the influence of positive APLA tests on therapy, screening can be justified in a many subjects. In relation to venous thromboembolism, all subjects with apparently spontaneous events should be considered for testing. The prevalence of positive tests is likely to be lower in thrombosis secondary to identified events such as surgery or trauma. Recurrent venous thromboembolism, even in the presence of other risk factors, may be an indication for testing for APLA. Subjects with stroke and those with peripheral arterial occlusive events occurring at a young age (for example less than 50 years) should be tested for APLA, especially when risk factors for atheromatous arterial disease are not prominent. The case can be made also for screening older subjects who are non-smokers and are not exhibiting other risk factors such as hypertension, diabetes mellitus or dyslipidaemia. Where recurrent arterial occlusive events occur despite antithrombotic prophylaxis, APS should be excluded. In subjects with SLE, APLA should be sought as part of the assessment of the autoantibody profile.
Because miscarriage is a common phenomenon, screening for APLA is not informative after a single event. In women with three or more consecutive pregnancy losses, testing for APLA should be part of the comprehensive investigation, including gynaecological, hormonal and chromosomal assessments. Unexplained loss of any morphologically normal fetus in the second or third trimester may be an indication for testing for APLA. Consideration should also be given to the possible diagnosis of APS in women with early severe pre-eclampsia or severe placental insufficiency in any pregnancy. Because maternal antiphospholipid antibodies may be downregulated during pregnancy, tests are best performed preconceptually or early in pregnancy when possible (Godeau et al, 1997).
1.4. Clinical manifestations of antiphospholipid syndrome
1.4.1. Clinical relevance of APLA
Although APLA were first described more than four decades ago, the mechanisms underlying their association with clinical events have not been completely defined.
A) APLA in healthy population
Estimates of the prevalence of APLA in healthy populations vary depending on the criteria used. It has been reported that ACA exist in approximately 5% of normal individuals, although less than 2% showed persistently elevated levels (Vila et al, 1994).
B) APLA in patients with no thrombosis or pregnancy morbidity, but with infections, drugs, auto-immune disorders
APLA are commonly found after certain acute or chronic infections. They develop in as many as 30% of children after viral infections. A high proportion of children and adults infected with mycobacteria, malaria, Q fever, hepatitis C, parvovirus B19, cytomegalovirus, etc. develop APLA (McNeil et al, 1991; Mengarelli et al, 2000). In HIV-positive patients, Constans et al (1998) found a 41% frequency of IgG APLA; IgM APLA were positive in 7% and IgG anti-b2GPI were rare (3-4%). In Geneva University Hospital, ACA were determined by an ELISA assay in 116 HIV-1-infected patients and positive test was found in 23.3% of the patients with a predominance of IgG ACA isotype (Bernard et al, 1990). Another study at Geneva University Hospital included 43 HIV-positive and 29 HIV-negative heavily transfused haemophiliacs. The presence of ACA was detected in 10 patients, all of them infected by HIV (Naimi et al, 1990).
The increase of APLA in patients infected with HIV may be due to disruption of the cell membranes which leads to the exposure of normally "hidden" PL during apoptosis (Clements et al, 1995). APLA from HIV-infected individuals tend to recognize various PL, most commonly PS (Petrovas et al, 1999). In addition, ACA in patients infected with HIV recognize oxidized CL more strongly than reduced CL. This finding, in conjunction with the increased oxidative stress found in these patients, may further explain the generation of APLA as a result of neoepitope formation by oxidized PL (Tzavara et al, 1997).
Some medications associated with the development of APLA include neuroleptics, quinidine and procainamide (Merrill et al, 1997). APLA could be induced also by phenothiazines, chlorthiazide, ethosuximide, oral contraceptives or alpha-interferon (Kutteh et al, 1997). The duration of APLA after infection or discontinuation of drug exposure is not well established, and the risk of thrombosis is variable.
LA have been identified in 10% to 20% of patients with well-established SLE, and ACA- in 30% to 50% of the individuals with SLE (Sammaritano et al, 1990) who present the majority of cases of secondary APS.
APLA also occur in patients with other autoimmune disorders like Sjögren's syndrome, mixed connective tissue disease, rheumatoid arthritis, systemic sclerosis, ankylosing spondylitis, vasculitis, idiopathic thrombocytopenic purpura, etc. APLA are found also in patients with diabetes mellitus, Crohn's disease and autoimmune thyroid disease. APLA can be detected in some patients with malignancies such as thymoma; carcinoma of the lung, kidney, ovary, cervix uteri, prostate; lymphoma, leukemia and various myeloproliferative disorders (Kutteh et al, 1997).
C) APLA in patients with thrombosis and/or pregnancy morbidity / primary APS /
According to the Sappporo's criteria for APS, all individuals with APS have by definition ACA and/or LA. Recent data show that LA are the strongest risk factor for thromboembolic events in patients with primary APS and secondary APS (Galli et al, 2000). No clear results have been reported showing that the measurement of ACA defines the patient's thrombotic risk. It has been found that ACA do not recognize anionic PL but are directed against plasma proteins bound to suitable anionic (PL and other) surfaces. Among them, b2GPI and Pt are the most common antigen targets. b2GPI is required by the great majority of ACA to react with CL in immunoassays (Galli et al, 1990). A recent study examined the distribution of APLAs among patients with SLE. The most prevalent APLA were found to be anti-b2GPI antibodies. They were observed in 36.8% of all patients with SLE, being present in 40.4% of SLE patients with secondary APS and 34.9% in SLE patients without clinical features of APS (Bruce et al, 2000).
1.4.2. Clinical criteria for APS
Preliminary criteria for APS were formulated during the International workshop in Sapporo, 1998. Definite APS is considered to be present in a given patient when at least one of the following clinical criteria exist:
-
Vascular thrombosis
One or more clinical episodes of arterial, venous, or small vessel thrombosis, in any tissue or organ. Thrombosis must be confirmed by imaging or doppler studies or histopathology, with the exception of superficial venous thrombosis. For histopathologic confirmation, thrombosis should be present without significant evidence of inflammation in the vessel wall. -
Pregnancy morbidity
- One or more unexplained deaths of a morphologically normal fetus at or beyond the 10th week of gestation, with normal foetal morphology documented by ultrasound or by direct examination of the fetus, or
- One or more premature births of a morphologically normal neonate at or before the 34th week of gestation because of severe preeclampsia or eclampsia, or severe placental insufficiency, or
- Three or more unexplained consecutive spontaneous abortions before the 10th week of gestation, with maternal anatomic or hormonal abnormalities and paternal and maternal chromosomal causes excluded.
A) Vascular thrombosis
A retrospective analysis of 100 patients with primary APS and secondary APS (Munoz-Rodriguez et al, 1999) reported one or more thrombotic episodes in 53% of the patients: 40% had venous thrombosis, 53% had arterial thrombosis, and 7% had combined arterial and vein thrombosis (Table 4).
Venous thrombosis
Venous thrombosis most commonly involve the deep venous system of the lower limbs and the pelvic region. Cervera et al (2002), studying 1000 patients with primary APS and secondary APS, showed that deep vein thrombosis is the most common clinical complication (38.9%). Deep vein thrombosis occurs most often in high-risk settings, such as pregnancy, prolonged immobilisation, or use of oral contraceptives. A pulmonary embolism was observed in 14%, and superficial thrombophlebitis in the leg in 11.7% of the patients during the evolution of their disease. Thrombi at unusual sites such as the hepatic, mesenteric, axillary, pulmonary, and renal veins, cerebral venous sinus, and inferior vena cava have also been reported.
Less than 1% of episodes of venous thromboembolism are fatal. Although significant morbidity from post-thrombophlebitic syndrome develops in around 30% of individuals with lower limb deep vein thrombosis, a significant proportion of these eventually becomes asymptomatic (Prandoni et al, 1999).
- Recurrence of thrombosis
Munoz-Rodriguez et al (1999) observed that in most of the patients with recurrent episodes of venous thrombosis, the thrombotic event was at the same site as the previous thrombosis. In recurrent venous thrombotic events in the legs, 45% of the individuals had recurrence in the same leg, 20.7 % in the controlateral and 30.5% in both legs (Margaglione et al, 1999). A prospective four years follow-up study compared the risk of recurrent venous thromboembolism in 412 patients with a first episode of venous thromboembolism, with or without APLA (Schulman et al, 1998). The risk of recurrence was found to be 29% in patients with ACA versus 14% in those without antibodies. The rates of recurrence were 10% per year in patients with ACA and 4% per year in those without such antibodies. The presence of elevated titers of ACA 6 months after an episode of venous thromboembolism was proposed as a predictor for an increased risk of recurrence of thrombosis and death. Recurrences of thrombosis were observed in 19% of the episodes treated with long-term oral anticoagulation with warfarin sodium or dicoumarol, in 42% treated prophylactically with aspirin, and in 91% in which anticoagulant/antiaggregant treatments were discontinued, which indicates the benefit from prolonged oral anticoagulation. Nojima et al (2001) found the presence of anti-protein S antibodies as a significant risk factor for venous thrombosis but not for arterial thrombosis.
Arterial thrombosis
Arterial thrombi can occur in any central or peripheral vessel. The most common arterial locations are the aorta, iliofemoral, cerebral, coronary and retinal arteries. The cerebral arterioles are the most common site of arterial thrombosis in APS. Peripheral arterial occlusion, with gangrene, is less common. In the study of Cervera et al (2002), 19.8% of the patients with APS had a stroke, and 11% had transient ischemic attack. Myocardial infarction was observed in 5.5% of the cases; arterial thrombosis of the legs has been found in 4.3%; from the ophtalmologic manifestations 5.4% of patients had amaurosis fugax and 1.5% had retinal artery thrombosis. Arterial thrombosis carries a much higher risk of morbidity and mortality due mainly to cerebral ischemia. More prolonged and intensive anticoagulant therapy is recommended.
- Recurrence of thrombosis
Krnic-Barrie et al (1997) observed recurrent arterial thrombosis in 55% of patients with a primary and in 38% of the individuals with a secondary APS. The recurrent arterial events were found mainly in the white race. Schulman et al (1998) reported 28% risk of recurrences of thrombosis in patients with a low positive titer of ACA (5 to 35 GPL units), and 38% risk of recurrences in patients with moderate or high titer (>35 GPL units). In Geneva Vogel et al (1991) investigated a group of 65 patients with SLE and found that half of patients had ACA associated with arterial thromboembolic events. Nojima et al (1997) studied the relationship between arterial or venous thrombosis and the levels of ACA and/or existence of LA. They observed that the prevalence of thrombosis was higher in a ACA and LA positive patients (84%) than in ACA only positive patients (16%), or LA only positive patients (9%). Furthermore, in these patients positive for the two tests, all patients with a high positive level of ACA had arterial thrombosis indicating that a high ACA activity combined with a LA positive result might be a risk factor for arterial thrombosis. In a recent study the authors reported that both anti-b2GPI and anti-Pt antibodies might be also significant risk factors for arterial thrombosis but not for venous thrombosis (Nojima et al, 2001).
Thrombotic complications associated with APLA
-
Cardiovascular manifestations
The most common cardiac lesions described in APS patients are heart valve lesions including thickening, stenosis, vegetation formation and mitral regurgitation. Valvular abnormalities were found in 36% of patients with primary APS and in 48% of patients with secondary APS (Nesher et al, 1997). Deposition of APLA in the subendothelial layer is suggested as the pathogenic mechanism (Durrani et al, 2002). Angina pectoris was found in 2.7% of the cases, myocardiopathy in 2.9% and subclavian vein thrombosis in 1.8% (Cervera et al, 2002). -
Central nervous system manifestations
The cerebral vasculature is a common site of arterial thrombosis in APS (Sheng et al, 1998). Multiple subcortical white-matter infarcts secondary to cerebral ischemia the most frequent finding on magnetic resonance imaging. Levine et al (1995), in their prospective study of 81 consecutive APS patients who developed cerebral ischemia, reported a recurrence rate of 31% during a follow-up period of 3 years. The median time to recurrence was 7.9 months; an IgG APL titer greater than 100 GPL was associated with an even shorter recurrence time. The reported frequency of neurological complications in APS patients was as follows: stroke in 42%, migraine headaches in 13-20%, epilepsy in 3-7%, multi-infarct dementia in 2%, chorea in 1%, and cerebral vein thrombosis in 0.7% (Munoz-Rodriguez et al, 1999; Cervera et al, 2002). Cerebral venous thrombosis was more common at a younger age and had a more extensive involvement in patients with APS (Carhuapoma et al, 1997). Seizures, transverse myelitis and chorea associated with APS were likely to be due to an interaction between central nervous system cellular elements and APL rather than thrombosis, although a firm link has not been established between these features and APS (Brey et al, 1998). -
Osteoarticular and cutaneous manifestations
As reported by Cervera et al (2002), the most common skin manifestations of APS are livedo reticularis (24.1%), leg ulcers (5.5%), pseudovasculitic lesions (3.9%) and digital gangrene (3.3%). They have been most commonly found on the extremities due to superficial venous thrombosis and thrombo-phlebitis. Avascular necrosis of bone was found in 2.4%, and arthritis in 27% of the patients with APS (Cervera et al, 2002).
B) Pregnancy morbidity
Pregnancy loss is a defining criterion for APS and occurs with a particularly high frequency in SLE patients. In addition to embryonic losses (before 10 weeks gestation) or foetal losses (after 10 weeks), APS is associated with a number of potential serious obstetric complications, including thrombosis, severe preeclampsia, utero-placental insufficiency, foetal distress and iatrogenic preterm birth (Table 5). These complications have significant maternal consequences, and they also may contribute to foetal loss. They may be associated also with other thrombophilic disorders such as factor V Leiden or G20210A mutations.
Cervera et al (2002) analysed 590 women with APS who had 1 or more pregnancies: 74% of them succeeded in having 1 or more live births. The most common obstetrical complications in the mothers were preeclampsia (9.5% of pregnant women), eclampsia (4.4%) and abruptio placentae (2%). The most common foetal complications were embryonic loss in 34.5% of the pregnancies, foetal loss in 16.9% of the pregnancies, and premature birth in 10.6% of life births.
In women with SLE, a previous adverse outcome was identified as the most important risk factor for another miscarriage in a subsequent pregnancy (Finazzi et al, 1996). Faden et al (1997) observed that the presence of anti-b2GPI antibodies correlates well with some obstetrical complications, mainly eclampsia and preeclampsia. Anti-IgM anti-b2GPI antibodies correlated well with a history of pregnancy loss (Forastiero et al, 1998).
In 1996, Oshiro et al performed a retrospective study of 366 women with two or more consecutive pregnancy losses, where 79 of them were noted to have LA or ACA, and 290 did not. Both groups had similar rates of pregnancy loss (84%). However, those with APS had 50% foetal deaths compared with 15% foetal deaths in those without APS. Approximately 80% of those with APS had at least one foetal death compared with less than 25% in those without APS. Branch et al (1997) studied 147 women with recurrent pregnancy loss, negative for LA and with medium-to-high levels of IgG ACA, 104 healthy, fertile controls of similar age and gravidity, and 43 women with well-characterized APS. Twenty-six (18%) women with recurrent pregnancy loss and nine (9%) controls tested positive (above the 99th percentile) for APLA. Sera from five (3.4%) women with recurrent pregnancy loss and four (3.8%) controls demonstrated binding to PL antigens other than CL.
1.4.3. Other manifestation of APS
A) Thrombocytopenia
Thrombocytopenia was observed in 30-52% of patients with APS (Munoz-Rodriguez et al, 1999; Cervera et al, 2002). It was found in 21% of the cases with primary APS and in 38% of cases with secondary APS. Thrombocytopenia in secondary APS patients was significantly associated with the presence of ACA at medium-high titer (Amoroso et al, 2003). It was usually mild (platelet count above 90 G/l) and, except in very severe cases, no bleeding was observed. Similarly to immune thrombocytopenias, pathogenic antibodies were directed towards epitopes on platelet membrane glycoproteins and were distinct from 'antiphospholipid' antibodies (Godeau et al, 1997). Galli et al (1994) measured in 68 patients with APLA also anti-GPIb/IX and GPIIb/IIIa IgG, directed against platelet membrane-associated glycoproteins. Increased plasma levels of these anti-glycoprotein antibodies were found in 40% of cases. Furthermore, APLA have been reported in around 30% of subjects with typical immune thrombocytopenias.
B) Atherosclerosis
Recent studies demonstrated that EC activation induced by APLA might accelerate atherosclerosis associated with APS (Ross et al, 1999). Several studies have shown a close link between an atherosclerotic process and APS (Harats et al, 1999; Bruce et al, 2000 (b)).
C) Catastrophic APS
Catastrophic APS is a rare, accelerated form of APS. The patients with catastrophic APS present with wide spread noninflammatory thrombi involving the kidneys, lungs, heart, gastrointestinal tract, liver, the central nervous system as well as adrenal glands, in various combinations. Individuals presented with a clinical picture of acute multi-organ thrombosis with a 60% mortality due to myocardial infarction, acute respiratory distress syndrome, renal failure, or stroke (Asherson et al, 1996; Triplett et al, 2000). In the study of Asherson et al (2001), a total of 80 patients with catastrophic APS were analyzed. The most important manifestations found at the onset of the episode were cardiopulmonary (25%), neurologic (22%), abdominal (22%), and renal (14%) with a very high mortality (48%).
1.5. Pathogenesis of the antiphospolipid syndrome
The pathogenesis of APS is not completely clear. One hypothesis is that the exposure of anionic PL during apoptosis may be the driving antigenic stimulus for the development of APLA (Pittoni et al, 1998). Another hypothesis is that viral or bacterial infections may initiate the production of APLA. Indeed Gharavi and Pierangeli (1998) observed that peptides from adenovirus 2, cytomegalovirus or from bacillus subtilis are homologous to PL-binding region of b2GPI. Some hypotheses to explain the pathogenic mechanisms in APS rely on the variety of effects of APLA. Indeed PL are involved in the hemostatic reactions and in biological processes in various manners and APLA have been shown to affect coagulation at different steps.
APLA have been shown to play a true causal role in development of thrombosis and obstetrical complications. Indeed immunization of mice with heterologous b2GPIð leads to the development of APLA with recurrent pregnancy loss and thromboembolic complications (Gharavi et al, 1998). Moreover mice infused with APLA developed significantly larger thrombi in femoral veins after experimental injury than mice infused with control antibodies (Pierangeli et al, 1996). Also, a monoclonal human ACA derived from a patient with APS promoted thrombosis in mice (Olee et al, 1996). Atherosclerosis in a susceptible mouse model (LDL-receptor knockout mice) was accelerated by immunization with human ACA from a patient with APS, providing additional evidence for a causal pathogenic effect (George et al, 1997). The recent characterization of chimpanzee b2GPI with the finding of a high prevalence of anti-b2GPI antibodies in these animals could propose a possibility of primate models for investigating APS (Sanghera et al, 2001). It is possible that APLA predispose to thrombosis either by causing cells to acquire a procoagulant phenotype or by inhibiting cell surface anticoagulant processes. Additional mechanisms may be involved in the pathogenesis of obstetrical complications.
1.5.1. Effects of APLA on the cells involved in hemostasis
APLA have been shown to interfere with different types of cells (Table 6) involved in hemostasis. Other cells such as fibroblasts may also play a role but they are not detailed in this review.
| Endothelial cells Platelets Mononuclear cells Polymorphonuclear cells |
A) Endothelial cells
Endothelium is a metabolically active interface between the blood and extravascular tissues. Resting EC exert anticoagulant and antithrombotic properties by preventing contact of blood with prothrombotic underlying tissues and by producing and presenting on the EC surface molecules that aid in this function (Pearson et al, 2000). When EC are activated by different stimulus such as inflammatory cytokines (TNFa, IL-1), bacterial lipopolysaccharide (endotoxin), viral infections or hypoxia, they express both procoagulant and proinflammatory properties. It is now known that APLA can bind to and also activate EC in a similar manner, thus provoking a procoagulant phenotype in EC. In the next paragraphs we will review the main properties of resting endothelium and the changes after its activation (Table 7).
Resting EC
Resting EC are mainly antithrombotic and thus thanks to different mechanisms. The main molecules are indicated in Table 7 (next page).
Prostacyclin (PGI 2) and nitric oxide (NO), also called endothelium-derived relaxing factor (EDRF), inhibit synergistically the platelet aggregation and also act as vasodilators. Besides vasodilatator and antiplatelet properties, nitric oxide has antiadhesive and antioxidative effects. The best-known antioxidant effect of nitric oxide is the impairment of lipid oxidation, mainly free fatty acids, phosphatidylcholine and low-density lipoprotein particles. In view of the proatherogenic effects of oxidized lipids, this antioxidative activity of nitric oxide is likely to be relevant (O'Donnell et al, 2001). The mechanism of the antiadhesive action of nitric oxide could also involve antioxidant effects. Indeed the increased leukocyte adhesion induced by inhibition of nitric oxide synthases was, at least partially, reversed by intracellular oxygen radical scavengers (Niu et al, 1994).
Both mediators, PGI2 and NO, are synthesized and released by EC locally and transiently in response to agonists molecules involved in the coagulation process (e.g. bradykinin and thrombin) or secreted by aggregating platelets (e.g. adenosine triphosphate and adenosine diphosphate). PGI2 and nitric oxide synthesis are each triggered by increases of intracellular calcium ion concentrations in EC. The elevation of calcium ion, required to activate fully the production of nitric oxide from its precursor arginine is lower than that needed to drive PGI2 synthesis (Gryglewski et al, 2001). As well as being triggered by platelet secretory products, PGI2 synthesis, unlike nitric oxide synthesis, can occur in an agonist/receptor-independent fashion when platelets aggregate. The activated platelets secrete PGH2 or arachidonate that is directly converted to PGI2 by endothelium.
Tissue factor pathway inhibitor (TFPI) is the physiological inhibitor of TF/factor VII complex, synthesized by EC and bound to the EC surface (Lupu et al, 1997).
Heparan-like sulfate proteoglycans are localized to the EC surface and they serve to accelerate the inhibition of thrombin by antithrombin.
Thrombomodulin (TM) is a cell surface proteoglycan produced by EC and expressed on their surface. TM binds thrombin and decreases its capacity to cleave fibrinogen but similarly increases its capacity to cleave circulating protein C (Esmon et al, 1995). On thrombin binding, the complex is endocytosed, thrombin is degraded, and TM is recycled to the cell surface.
Endothelial protein C receptor, expressed by EC, enhances the protein C activation by the thrombin-TM complex (Laszik et al, 1997; Esmon et al, 2003).
Protein S, synthesized and secreted by EC, is a co-factor that promotes protein C anticoagulant pathway.
Tissue-type plasminogen activator (t-PA)is a fibrinolytic mediator, secreted from Weibel-Palade bodies in response to thrombin (Emeis et al, 1997; Huber et al, 2001; Rosnoblet et al, 1999). Classically, t-PA is activated by binding to fibrin, hence localizing plasmin generation to the site of a clot. In addition, the EC surface possesses several binding sites for plasminogen and a specific t-PA receptor, which leads to local plasmin generation at the EC surface (Hajjar et al, 1995).
Annexin V, expressed on EC, binds with high affinity to the surface PL of both quiescent and activated EC and inhibits the procoagulant reactions.
Ecto-adenosine diphosphate and adenosine triphosphate receptors are expressed on EC surface. Adenosine diphosphate receptors induce a response on EC initiating PGI2 and nitric oxide synthesis. The major pathway responsible for ending the pro-aggregatory action of adenosine diphosphate is its sequential dephosphorylation to adenosine monophosphate and then adenonosine (an inhibitor of platelet aggregation), which is due to ectonucleotidase enzymes at EC surface (Zimmerman et al, 1998). Adenosine diphosphatase, secreted from activated platelets degrades adenosine diphosphate and thereby limits the effect of platelet released adenosine diphosphate.
Activated EC
Endothelial activation leads to loss of anticoagulant properties (Table 7) and is characterised by several modifications.
Diminution of NO
Recent studies in hypercholesterolaemic animals and humans show that the deficiency in agonist-induced nitric oxide synthesis can be improved by elevating the circulating levels of arginine. This suggests that the intrinsic levels of nitric oxide synthase are preserved but that some aspects of the coupling between agonist receptor and nitric oxide synthesis are disturbed (Maxwell et al, 1998). In contrast with nitric oxide decrease, PGI2 release is enhanced in patients with atherosclerosis. This has been attributed as a consequence of excessive platelet reactivity.
Tissue factor expression
Tissue factor is a transmembrane protein expressed by stimulated EC. It is the physiological trigger of normal coagulation and a major initiator of clotting in thrombotic disease (Figure 5). Tissue factor initiates the extrinsic pathway of coagulation by serving as a cofactor and receptor for factor VIIa to efficiently cleave its substrates, factor IX and factor X, to their active forms (Roubey et al, 2000).
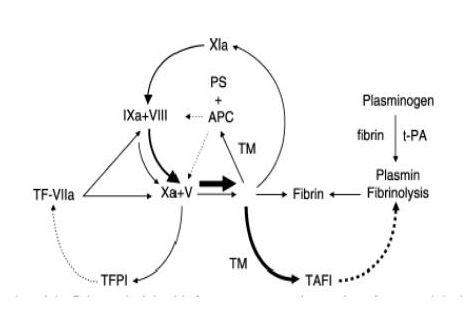
|
TF-VIIa: tissue factor-FVIIa complex TFPI: tissue factor pathway inhibitor APC: activated protein C PS: protein S T: thrombomodulin TAFI: thrombin-activatable fibrinolysis inhibitor t-PA: tissue-type plasminogen activator |
An uninterrupted line indicates activation, while an interrupted line indicates inactivation. The uninterrupted line between Xa and TFPI indicates that FXa has to form a complex with TFPI, and that this complex then inhibits TF-FVIIa.
Increase in cell surface anionic PL
Activated EC have increased exposure of surface anionic PL, which are cofactors for the coagulation system.
Diminution of TM
Activated EC down-regulate the synthesis and cell surface expression of TM and consequently reduce the formation of the thrombin-TM complex, thereby limiting thrombin generation (Laszik et al, 2001).
Secretion of plasminogen activator inhibitor type I (PAI-I)
PAI-I is synthesized and secreted by activated EC. It is the major plasma inhibitor of t-PA, involved in the regulation of fibrinolysis, degradation of the extracellular matrix and angiogenesis.
Release of Von Willebrand factor (vWF)
Activated EC release vWF from storage granules (Weibel-Palade bodies) in response to thrombin, which participates in platelet adhesion.
Cell surface protease receptors expression
Activated EC serve as a site of protease by expressing cell surface protease receptors. Thus EC facilitate the formation of enzyme complexes involved in the regulation of coagulation and fibrinolysis.
Increased adhesion molecule expression
Activated EC express adhesion molecules for leucocytes such as E-selectin, intracellular adhesion molecule (ICAM-1 and -2), vascular cell adhesion molecule (VCAM-1).
-
Effects of APLA on endothelial cells
It has been hypothesized that APLA bind to antigens such as PS, TM and heparan proteoglycan on EC surfaces (Pierangeli et al, 1999) and that b2GPI could be a cofactor facilitating this interaction with EC (Del Papa et al, 1997; Dueymes et al, 1996). Indeed EC activation by APLA is associated with increased expression of adhesion molecules, increased synthesis and secretion of proinflammatory cytokines, tissue factor expression, increased endothelin-1, induction of apoptosis, and EC migration.
Increased expression of adhesion molecules
Activated EC express surface E-selectin, VCAM-1, and ICAM-1 leading to increased monocytes and leucocytes adhesion. (Simantov et al, 1995; Meroni et al, 2001). Pierangeli et al (2000) demonstrated that in a pinch-induced thrombosis model APLA enhance leukocyte adhesion and increase thrombosis. The authors analyzed in vivo leukocyte adhesion to endothelium in venules of exposed murine cremaster muscle. The thrombogenic effects of APLA were reduced in transgenic ICAM-1-deficient mice, ICAM-1/P-selectin-deficient mice and in mice infused with anti-VCAM-1 antibodies.
Increased synthesis and secretion of proinflammatory cytokines
APLA increase EC synthesis and secretion of the proinflammatory cytokines IL-1 and IL-6. This could further contribute to cell activation in an autocrine manner because specific antagonists, such as IL-1 receptor antagonist, can inhibit the process (Del Papa et al, 1997; Meroni et al, 2000). Thus anti-b2GPI antibodies can induce an EC activation either directly or by a cytokine autocrine loop.
Tissue factor expression
It has been demonstrated that incubation of cultured EC with anti-b2GPI antibodies result in an increased production of TF, further supporting the hypothesis that APLA are procoagulant triggers (Branch et al, 1993 ; Kornberg et al, 2000; Dunoyer-Geindre et al, 2001). Amengual et al (1998) demonstrated by reverse-transcription polymerase chain reaction that human monoclonal anti-b2GPI antibodies upregulate tissue factor mRNA on HUVEC, suggesting that the tissue factor pathway be implicated in the pathogenesis of APLA related thrombosis.
Increased endothelin-1 (ET-1)
Significantly increased plasma levels of ET-1, the most potent endothelium derived contracting factor, were found in patients with APS and arterial thombosis (Atsumi et al, 1998). In vitro incubation of EC with human monoclonal anti-b2GPI antibodies was shown to upregulate the expression of preproendothelin-1 mRNA.
Induction of apoptosis
A subset of APLA that recognizes annexin V induces apoptosis in EC (Nakamura et al, 1998; Pittoni et al, 1998). In vivo, apoptosis of EC would lead to de-endothelialization and exposure of the thrombogenic subendothelium (Bombeli et al, 1999). It is hypothesized that APLA could also displace annexin V that covers the anionic PL on EC membranes, thus increasing the net quantity of thrombogenic PL exposed their procoagulant phenotype (Rand et al, 2000).
EC migration
APLA may also interfere with EC migration. This activity could potentially interfere with re-endothelialization and prolong the exposure of the thrombogenic subendothelium (Lanir et al, 1998).
B) Platelets
Platelet binding to activated EC is the next phase in the process of thrombus formation although the precise mechanisms that mediate EC-platelet interaction are not completely clear. It was demonstrated that activated platelets are present in patients with APS but whether platelet activation in patients with APLA is a direct result of APLA or other autoantibodies or a consequence of vascular injury is uncertain (Emmi et al, 1997). A recent study found that APLA in APS have antiplatelet reactivity but there was no evidence for associated direct platelet-activating ability (Ford et al, 1998). Galli et al (1996) reported that the evidence that human APLA either bind to or activate platelets is still uncertain. Lackner et al (2000) also showed that two human monoclonal APLA had no effect on platelets as determined by flow cytometric analysis of CD62P, CD41, CD42b expression and fibrinogen binding with and without previous activation with adenosine diphosphate or thrombin receptor activating peptide (TRAP-6). One possible explanation for these observations might be the presence of specific antiplatelet autoantibodies that coexist with APLA in patients with APS (Reverter et al, 2000). However, some investigators have shown that APLA may induce platelet activation and aggregation in the presence of low concentrations of agonists such as thrombin, adenosine diphosphate or collagen (Martinuzzo et al, 1993; Campbell et al, 1995; Nojima et al, 1999). A correlation was found between the IgG level of ACA and the CD62-positive platelet percentage in patients with primary APS and, more significantly, in the patients with primary APS and neurological disorders.
APLA-containing plasma promoted platelet aggregation in a perfusion model (Escolar et al, 1992). Wiener et al (2001) suggested that the platelet aggregation in APS is induced by an APLA-complex present in patients' plasma. The initial trigger in this thrombotic process is calcium independent, but probably is followed by release, recruitment, and ultimately fibrin formation by the usual metabolic calcium-dependent fibrinogen binding pathway. Potential targets for APLA on platelets include platelet-activating factor (Barquinero et al, 1994), PS (Vazquez-Mellado et al, 1994; Campbell et al, 1995), and platelet glycoprotein IIIa (Tokita et al, 1996). Recently Ferro et al (1999) characterized 11-dehydro-TXB2 as a sensitive marker of platelet activation, and found it significantly higher in patients with SLE and APLA. A statistically significant correlation was found between plasma levels of vWF and tPA and excretion of this thromboxane metabolite. In a recent study, levels of platelet activation were investigated in 20 patients with primary APS and 30 SLE patients (14 of whom had secondary APS) by measuring CD63 expression on platelets and soluble P-selectin levels. Platelet CD63 expression and soluble P-selectin levels were significantly higher in patients with primary APS and SLE patients with/without APS than normal controls (Joseph et al, 2001). Robbins et al (1998) hypothesized that APLA/b2GPI complexes could activate platelets to produce thromboxane A2 (a proaggregatory prostanoid) which could contribute to the prothrombotic state found in patients with APS. Shechter et al (1999) found that platelet serotonin concentration in APS patients was significantly lower than that found in the platelets of normal controls but the reasons of low serotonin levels are still not clear.
Thrombocytopenia is a common finding in patients with APS and a potential association with platelet activation could exist. It was hypothesized that thrombocytopenia is due to platelet activation and consumption of platelets on the damaged vascular endothelium (Walenga et al, 1999). George et al (1999) studied 38 SLE patients and found in 26.3% of them high levels of b2GPI containing immune-complexes. There was a positive correlation between b2GPI/ immune-complexes levels and the occurrence of thrombocytopenia.
C) Monocytes
Monocytes are implicated in the pathogenesis of APLA related thrombosis, mainly by stimulated cell surface expression of tissue factor (Kornberg et al, 1994; Cuadrado et al, 1997). Amengual et al (1998) revealed by flow-cytometry that monocytes from a healthy donor displayed higher tissue factor antigen expression when incubated in the presence of APS plasmas than with control plasmas. Stimulation of monocytes from APS patients with b2GPI induced substantial monocyte TF, whereas no induction was observed with cells from patients having APLA without APS. Tissue factor induction on monocytes by b2GPI was dose dependent and required circulating type 1 (Th1) CD4+ T lymphocytes and class II Major Histocompatibility Complex (MHC) molecules (Visvanathan et al, 2000). The authors previously reported that at least 44% of patients with APS possess Th1 CD4+ T cells that proliferate and secrete IFN-g when stimulated with b2GPI in vitro (Visvanathan et al, 1999). F(ab)2 fragments of ACA have been reported to induce monocyte tissue factor expression as well, suggesting the contribution of an Fc-independent component to the mechanism of antibody-mediated tissue factor activity in APS (Kornberg, 1994). Increased levels of tissue factor mRNA have been found in the majority of mononuclear cell samples from patients with APS (Dobado-Berrios et al, 1999).
D) Polymorphonuclear cells
Arvieux et al (1995) investigated the ability of six murine monoclonal antibodies to b2GPI to induce polymorphonuclear cell functional responses. The six monoclonal antibodies tested in combination with b2GPI led to a concentration-dependent activation of human polymorphonuclear cells. The activation of polymorphonuclear cells was estimated by their granule release, H2O2 production, and cytosolic Ca2+ increase. The results showed that the process of polymorphonuclear cell activation depends on monoclonal antibody binding to these cells through both Fab (via b2GPI) and Fc domains.
1.5.2. Effects of APLA on coagulation
The effects of APLA on haemostatic reactions are shown on Table 8.
A) Interference of APLA with the components of the protein C/S pathway
The activation of the coagulation and the protein C pathway are shown in Figures 6, 7 and 8. Once activated by the thrombin-TM complex on the surface of EC, activated protein C (APC) exerts an inhibitory effect by cleavage of the factor Va and VIIIa; protein S and factor V are required as cofactors for the APC activity in vivo (Dalhback et al, 1993).
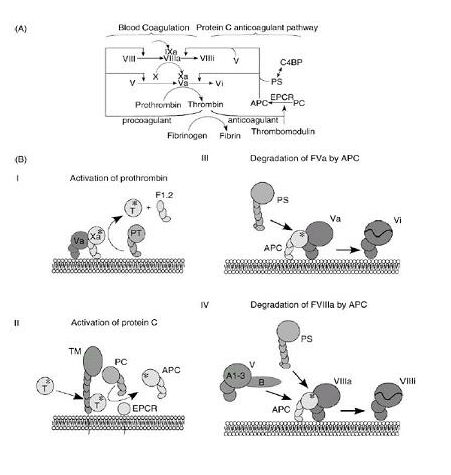
Section A demonstrates an incomplete scheme of blood coagulation reactions together with the balancing anticoagulant reactions of the protein C pathway. In section B (I-IV), the membrane-bound molecular events of selected reactions are shown in cartoon-like fashion. I: activation of prothrombin (PT) to thrombin (T), a reaction that also generates the F1.2 prothrombin fragment. II: thrombomodulin (TM) and the endothelial protein C receptor (EPCR) are proteins that span the membrane. The role of EPCR is not fully understood, but it has been shown to be able to bind the Gla-domain of protein C, which results in stimulation of protein C activation. III: the degradation of FVa by APC is enhanced by protein S (PS). IV: degradation of FVIIIa by APC is stimulated by the synergistic cofactor activity of protein S and factor V. The large B domain that protrudes from the triangularly arranged A1-A3 domains of FV is in the linear sequence located between A2 and A3 domains.
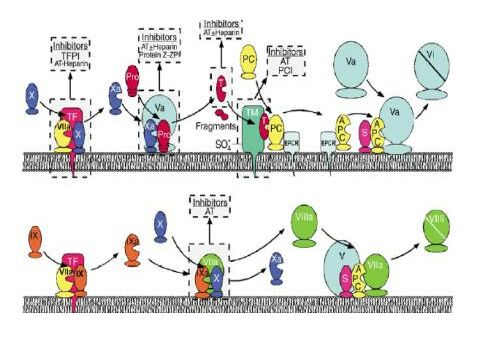
Inhibitors that control coagulation are shown in gray boxes above the complex or factor they regulate (AT: antithrombin, ZPI: protein Z-dependent protease inhibitor, PCI: protein C inhibitor). Top panel: FVIIa binds to tissue factor (TF) to activate FX, generatingFXa. FXa then binds to FVa. The complex of FXa-FV converts prothrombin to thrombin (T). Thrombin can then either bind to TM or carry out procoagulant reactions like fibrin formation or platelet activation. When bound to TM, thrombin can activate protein C (PC) to APC. This process is enhanced when protein C is bound to the EPCR. APC bound to EPCR cleaves substrates other than FVa. APC dissociates from EPCR and can then interact with protein S to inactivate FVa. Bottom panel: The FIXa-FVIIIa complex is inactivated by APC. In this case, FV participates with APC and protein S in the inactivation of FVIIIa. For simplicity, the activation of FVII, FV and FVIII are not shown.

Protein C activation takes place by way of interaction between the thrombomodulin-thrombin complex and the endothelial protein C receptor. Activated protein C, together with its cofactor, protein S, inactivates factors V and VIII to provide negative feedback to the generation of thrombin. Complex 1 comprises TF and coagulation factors VII, IX and X; complex 2 comprises factors IX and X and cofactor VIII; and complex 3 comprises factor X, prothrombin and cofactor V.
Experimental findings (de Groot et al, 1996) consistently showed that APLA may interfere with protein C axis in multiple ways as described below.
- Inhibition of APC anticoagulant pathway, directly or via its cofactor protein
Several recent studies proposed that APLA cause direct inhibition of the APC pathway and investigated the correlation of APLA specificity and APC pathway inhibition. Bokarewa et al (1994) studied the effect of 38 IgG fractions with either ACA alone or both ACA and LA on the response to APC. Five of eight IgG fractions with LA activity showed a tendency to reduce the effect of APC in the aPTT system and to simulate the activated protein C resistance (APC-R) phenomenon. No correlation was found between protein C activity and ACA levels or the extent of clotting time prolongation. In addition, Nojima et al (2002) found that the co-existence of anti-Pt antibodies and LA activity was a significant risk factor in the pathogenesis of APC-R in patients with SLE. Mali et al (2001) reported similar results from 59 unselected children with SLE showing that acquired APC-R was significantly associated with the presence of LA but not ACA. They proposed that acquired APC-R could be a marker identifying LA-positive patients at high risk of thrombosis. Controversially, Martinuzzo et al (1996) found that the acquired APC-R in patients with APS seems to be associated with ACA and anti-b2GPI rather than an in vitro interference by LA. Atsumi et al (1998) demonstrated in vitro that ACA antibodies (not anti-protein C autoantibodies) can bind protein C via b2GPI, and suggested that protein C could be a target of APLA by making a complex of protein C with ACA and b2GPI, leading to protein C dysfunction.
In addition, patients with APS were often found to have protein S deficiency. Atsumi et al (1997) demonstrated monoclonal ACA binding to protein S in presence of a combination of b2GPI and CL, with a consequent increase of the affinity of C4b-binding protein for protein S. Protein S could represent one of the targets for ACA when combined with b2GPI and CL, thus explaining the acquired free protein S deficiency and the attendant risk of thrombosis in patients with ACA. To explore the coagulation/fibrinolytic balance and its relation with free protein S, Ames et al (1996) carried out a cross-sectional study on 18 thrombotic patients with primary APS and 18 apparently healthy subjects with persistence of idiopathic APLA. Low free protein S was found in all non-thrombotic and in 90% of thrombotic patients with defective fibrinolysis. The data are consistent with increased thrombin generation and accelerated fibrin turnover and fibrinolysis abnormalities in asymptomatic subjects with APLA. It indicates also a possible central role for acquired free protein S deficiency in the thrombotic tendency of APS patients.
- Interference with the activation of protein C by the thrombomodulin-thrombin complex
Oosting et al (1993) showed that the anti-TM antibodies inhibiting TM and subsequent protein C activation are directed against the regions containing the epidermal growth factor (EGF) domains in SLE patients with a history of thrombotic complications. When TM was incorporated in PL vesicles, no inhibition by these anti-TM antibodies could be demonstrated. In addition, anti-TM antibodies could not inhibit protein C activation mediated by cultured EC. A conclusion was made that anti-TM antibodies inhibit only soluble TM.
Carson et al (2000) tested 58 patients with LA and found anti-TM antibodies in 30% of the cases. Similar antibodies were found in only 2% of 201 normal controls. Three IgG fractions of the 6 purified IgG from patients with anti-TM antibodies inhibited protein C activation from 40% to 70% compared to no inhibition in 7 healthy controls.
- Inhibition of thrombin formation
Prothrombinase, which consists of factor Xa, factor Va, Ca2+, and PL, converts Pt to thrombin. Thrombin is the final protease of the coagulation system and, in turn, activates protein C in the presence of thrombomodulin on endothelial cells. APC completes a negative feedback loop in the blood coagulation pathway by degrading factor Va and factor VIIIa and thereby inhibits prothrombinase activity (Stenflo et al, 1884). Using chromogenic substance assays and human IgM monoclonal ACA, Ieko et al (1999) found that these monoclonal ACA inhibited both thrombin generation and APC activity. Thrombin generation without APC was inhibited as well by adding these antibodies. This inhibition required the presence of b2GPI and confirmed the data presented previously by Mori et al (1996).
- Binding to coagulation-activated cofactors Va and VIIIa in a manner that protects them from proteolysis by APC
Borrell et al (1992) studied the effect of purified IgM and IgG from 21 patients with APLA on factor Va degradation by APC on HUVEC. Thirteen of 14 IgM and 8 of 10 IgG from patients showed an inhibitory effect on factor Va degradation by APC when compared with control Ig. Oosting et al (1993) investigated the effect of 30 IgG fractions APLA on APC-mediated factor Va inactivation in the absence and presence of protein S. Three IgG fractions inhibited APC-mediated factor Va inactivation independent of protein S and four IgG fractions APLA inhibited in the presence of protein S. The anticoagulant response of purified APC, added to LA-containing plasmas of 46 patients, was measured through the amount of factor VIII inactivation and an acquired APC dysfunction was found (Potzsch et al, 1995). Thirteen of 14 patients with recurrent thrombotic events and 10 of 19 patients with one single episode of thrombosis showed an abnormal APC response. In contrast, among 13 patients with LA without symptoms, only one showed an abnormal APC response.
A subset of APLA reactive with PE was found particularly active in inhibiting the ability of PE to promote APC-mediated inactivation of factor Va. This effect correlated poorly with LA activity and it was suggested that this 'acquired' APC resistance might be a risk factor for thrombosis even in the absence of LA (Smirnov et al, 1995).
APLA also interfered with the fibrinolytic pathway through TM inducible fibrinolysis inhibitor and by increasing PAI-1 activity. These findings suggested that the impairment of fibrinolytic activity by APLA might be one of the causes of thrombophilic diathesis in APS (Ieko et al, 1999).
- Interference of APLA with the components of intrinsic pathway
Recent studies reported the presence of antibodies to factor XII in a significant number of patients with APS suggesting that their presence might lead to acquired factor XII deficiency (Jones et al, 2000). Jone et al (2002) reported that, although factor XII is a member of the family of proteins which include plasminogen and Pt, antibodies to factor XII in patients with APS appear to be distinct from antibodies to Pt. Sugi et al (2001) reported that certain anti-PE antibodies are not specific for PE, but recognize the contact proteins factor XI and prekallikrein independently or in combination with high molecular weight kininogen. The contact proteins such as high molecular weight kininogen, prekallikrein and factor XII have anticoagulant and profibrinolytic functions and their deficiency was found to be associated with recurrent thrombosis. Several studies confirmed the presence of autoantibodies to the contact proteins in patients with SLE, thrombosis, and recurrent pregnancy loss.
- Inhibition of antithrombin activity by APLA
The formation of thrombin-antithrombin complexes (TAT) in APS was impaired. Ieko et al (2000) did not find an increase in level of TAT in APS, while the level of prothrombin fragment 1+2 increased. Therefore, free thrombin present in patient blood might contribute to thrombosis in APS.
It was demonstrated that at least some APLA cross-react with highly polyanionic heparin and heparinoid molecules and inhibit the acceleration of antithrombin activity (Shibata et al, 1994). Purified IgG from seven patients with APS were reactive with heparin by ELISA, whereas none of five controls had antiheparin reactivity. Specificity studies showed that APS IgG antiheparin antibodies were specifically reactive with a disaccharide present in the heparin pentasaccharide that binds antithrombin. Furthermore, these antibodies inhibited heparin-accelerated formation of antithrombin-thrombin complexes.
B) Impairment of fibrinolysis by APLA
Ieko et al (2000) investigated the effect of both b2GPI and APLA on the activity of extrinsic fibrinolysis. The authors found that b2GPI, without PAI-1, did not affect t-PA activity in a chromogenic assay. When PAI-1 was added to t-PA, the remaining t-PA activity was increased up to 60% by the addition of b2GPI. The effect of b2GPI did not require PL. Thus, b2GPI might protect t-PA activity from the inhibition by PAI-1. When monoclonal ACA were further added to the mixture with a diluted PL, the remaining t-PA activity decreased to 50 and 80%. Monoclonal ACA appeared to inhibit the effect of b2GPI and to elevate PAI-1 activity. Thus, the impairment of fibrinolytic activity by ACA might be one of reasons for the increased incidence of thrombosis in patients with ACA.
C) Additional effects of APLA
As PL bear structural resemblance to low density lipoprotein (LDL), several studies (Horkko et al, 1997) showed that APLA might show cross reactivity against oxidized LDL. Oxidized low-density lipoprotein is implicated in atherosclerosis by influencing foam cell formation and cell cytotoxicity. The production of anti-oxidized low-density lipoprotein antibodies results in the formation of immune complexes which are taken up at enhanced rate by macrophages, leading to foam cell formation. George et al (1997) demonstrated in mice immunized with ACA that developed APS, that infusion with oxidized low-density lipoproteins aggravated the manifestations of experimental APS. The authors suggested that cross-reactivity of oxidized low-density lipoproteins with PL might lead to significantly more severe form of APS.
In summary, although many studies were and are still performed to determine how APLA might increase thrombus formation in vitro, their mechanism of action remains unclear. Several studies have proposed that APLA activation of EC, platelets, and monocytes, as well as their effects on fibrinolysis and proteins C and S, may contribute to the prothrombotic state in APS. The large variety of proposed mechanisms makes it difficult to identify either the main primary mechanism. It is possible also that different mechanisms exist or co-exist among patients and even in one single patient.
1.5.3. Potential mechanisms involved in APLA-associated foetal loss
A relation between APLA and pregnancy loss has been formally recognized for almost 30 years (Nilsson et al, 1975). It is now accepted that APLA can lead to foetal loss and probably to recurrent preembryonic and embryonic loss. Studies have reported the presence of thrombi in placenta of patients with APLA-associated foetal loss, as well as other abnormalities, such as decreases in vasculo-syncytial membranes, villous fibrosis and hypovascular villi, and an increase in syncytial knots in placenta from 30% to 50% of these patients (Levy et al, 1998). Donohoe et al (1999) proposed that one of the mechanisms in APS associated obstetrical complications may involve the binding of APLA directly to the placenta where they may initiate placental thrombosis and infarction. By immunofluorescence techniques the authors detected human APLA binding to human placenta. Heterogeneous binding to normal term placenta, involving the trophoblast microvillous surface, the stromal and the peri-vascular regions was demonstrated by affinity purified APLA from five out of six patients.
However, the frequency and extent of thrombosis might not be sufficient to explain the high incidence of foetal loss in patients with APLA. Autoantibodies from sera of primary and secondary APS patients were found to affect reproductive outcome in pregnant mice in vivo (Matalon et al, 2002). Purified IgG from women with APS and recurrent pregnancy loss was injected to mice and affected directly the embryo and yolk sac reducing their growth. The purified IgG ACA reduced yolk sac and embryonic growth more than sera negative for these antibodies and caused foetal resorptions and growth retardation as well. In their study, Blank et al (1999) converted the anti-b2GPI monoclonal antibody into single-chain and replaced the H and L chains between the pathogenic and non-pathogenic single-chain anti-b2GPI. They demonstrated that single-chain of pathogenic anti-b2GPI are capable of inducing the same clinical manifestations as the whole antibody molecule in actively immunized mice. Elevated titers of mice ACA and anti-b2GPI, associated with LA activity, thrombocytopenia, prolonged aPTT and a high percentage of foetal resorptions were detected. Vogt et al (1997) observed that monoclonal anti-PS antibodies bind to choriocarcinoma cells as well as to trophoblast cells in histologic sections and, like anti-b2GPI antibodies, may displace annexin V from trophoblasts. Rand et al (1997, 1998) found that IgG APLA reduced the levels of syncytiotrophoblast apical membrane-associated annexin V in placental villi and the release of annexin V into surrounding media. In addition, trophoblasts and endothelial cells exposed to IgG APLA had significantly faster coagulation times. The authors proposed that the reduction of this anticoagulant protein at the maternal-foetal interface might account for the pregnancy loss in patients with APS.
The effect of APLA on several non coagulation-related placental functions, such as the secretion of human chorion gonadotropin, was also considered (Gleicher et al, 1992). An improved reproductive outcome in animal models of APLA-associated foetal loss in response to nonanticoagulant therapies, such as interleukin-3 or ciprofloxacin, suggested that mechanisms other than or in addition to thrombosis might contribute to placental dysfunction during APLA pregnancy (Blank et al, 1998). Holers et al (2002) found that APLA induce complement activation in the placenta resulting in placental injury, foetal loss and growth retardation. They used a murine model of APS in which pregnant mice were injected with human IgG containing APLA. The authors found that inhibition of the complement cascade in vivo, using the C3 convertase inhibitor complement receptor 1-related gene/protein y (Crry)-Ig, blocks foetal loss and growth retardation. Furthermore, mice deficient in complement C3 were resistant to foetal injury induced by APLA.
In summary, several mechanisms are involved in APS associated pregnancy loss. They result in uteroplacental insufficiency with usually multiple placental thromboses and infarcts, provoked by the hypercoagulable properties of APLA.
1.6. Management of the antiphospholipid syndrome
1.6.1. Treatment and prevention of venous thromboembolism
The initial management of an acute event, with intravenous monitored unfractionated or subcutaneous low molecular weight heparin is not really influenced by the diagnosis of APS. Warfarin therapy should be instituted in the usual way, with a target international normalized ratio (INR) of 2.5 (range 2.0-3.0). The duration of treatment should be determined on an individual basis, taking into account the presence of additional risk factors, the severity of the presenting event and the particular risk of bleeding on warfarin (Prandoni et al, 1996; Prandoni et al, 1999). Recent studies found that short-term warfarin therapy was not sufficient for patients with APS due to the high risk of recurrence of venous thromboembolism. In these cases the use of aspirin did not offer a clear additional benefit. Schulman et al (1998) demonstrated the beneficial effects of prolonged oral anticoagulation on patients with ACA after an episode of venous thromboembolism. Kearon et al (1999), in their study of 162 patients with a first episode of idiopathic venous thromboembolism, proposed also that these patients should be treated with anticoagulant agents for longer than usual because of the increased risk of recurrence of thrombosis. Some other studies (Brunner et al, 2002) reported that patients with APS and recurrent venous thrombosis should benefit from long term warfarin at higher intensity (INR 3.0-3.5). Although the risk-benefit ratio of long-term anticoagulation has not yet been clearly assessed, it is now proposed that for the majority of subjects having persistent APLA, a lifelong anticoagulant treatment (INR 2.0-3.0) should be provided after the first venous thromboembolism (Bauer et al, 2003).
1.6.2. Treatment and prevention of arterial thrombosis
Because of the high risk of recurrence and likelihood of consequent permanent disability or death, stroke due to cerebral infarction in patients with APS should be treated with long-term oral anticoagulant therapy, target INR 2.5 (range 2.0-3.0). Extracerebral arterial thromboembolic manifestations of APS will also warrant consideration of continuation of long-term anticoagulation with warfarin in many instances. Higher-intensity anticoagulation had been recommended and may be appropriate in some cases (Guidelines on oral anticoagulation, 1998), but results of prospective studies are required before the use of a target INR of 3.0 and more can be unequivocally supported.
The use of aspirin and/or hydroxychloroquine may be protective against thrombosis in asymptomatic APLA-positive individuals (Erkan et al, 2002). The addition of aspirin 100 mg to antivitamin K (INR 2.0-3.0) is now discussed in case of arterial thrombosis.
1.6.3. Prevention of pregnancy failure
In women with APS and a history of pregnancy complications, there is a particular need for a close collaboration between specialists. A variety of treatments including corticosteroids, low-dose aspirin, heparin and immunoglobulins have been used either as single agents or in combination in an attempt to improve the rate of live births in women with APLA (Kutteh et al, 1996). Available data are limited by the small number of patients in each study and by the lack of standardization of laboratory assays used to detect APLA. The use of corticosteroids in pregnancy is associated with significant maternal and foetal morbidity and should be avoided (Laskin et al, 1997). In their randomized controlled clinical trial, Rai et al (1997) reported that treatment with low-dose aspirin and heparin is applicable to women with a history of recurrent miscarriage associated with persistent APLA. They recommended treatment with aspirin, 75 mg/d, to be started as soon as the urine pregnancy test becomes positive. Because the majority of miscarriages occur before 14 weeks of gestation, the authors proposed a low-dose heparin treatment to be commenced when foetal heart activity is seen on ultrasonography. In the same study (Rai et al, 1997), unfractionated heparin (5000 IU) was administered twice daily subcutaneously. At present, although largely used, no low molecular weight heparin preparation is licensed for use in pregnancy in Switzerland. However many studies indicate that low molecular weight heparin preparations are safe alternatives to unfractionated heparin as an anticoagulant during pregnancy (Sanson et al, 1999). Concerning the potential risk of heparin-induced osteopenia, discontinuation of heparin therapy at 34 weeks gestation seems reasonable in women with early pregnancy loss and no history of thrombosis. When late pregnancy complications have occurred previously, continuation of antithrombotic therapy to delivery is reasonable and postpartum thromboprophylaxis is indicated in women with a history of thrombosis. Delivery by caesarean section carries an additional thrombotic risk and it is an indication for perioperative thromboprophylaxis. Although combination treatment with aspirin and heparin leads to a high live birth rate among women with recurrent miscarriage and APLA, successful pregnancies may be complicated by foetal growth retardation, gestational hypertension and premature delivery (Backos et al, 1999). In summary, optimal management, usually with low-molecular weight and aspirin, requires close collaboration between the haematologist and the obstetrician as well as facilities to enable appropriate clinical monitoring and laboratory testing.
A) Primary thromboprophylaxis in case of APLA
Controversial data exist regarding the prophylactic treatment of patients positive for APLA and without history of thrombosis. Incidental detection of low titers of APLA carries a minimal risk of thrombosis. However, there is convincing evidence to suggest a significant risk associated with the presence of medium to high levels of APLA. Prophylactic therapy is not accepted in clinical practice at present, although there is increasing evidence of the risks of high levels of APLA (Finazzi et al, 1996; Khamashta et al, 1999). Recent guidelines on the investigation and management of APS by Greaves et al (2000) suggested the prophylactic use of low-dose aspirin, which was shown to be protective of deep vein thrombosis in high risk groups (Rai et al, 1997). Prophylactic low-intensity oral anticoagulation with warfarin and low-dose aspirin has been shown to significantly reduce the incidence of ischemic heart disease in patients with APS (General Practice Research, 1998). A decision analysis study of patients with SLE, with and without APLA, also supports the prophylactic use of aspirin (Wahl et al, 2000).
B) Thromboprophylaxis with statins in case of APLA
Recent literature reported that statins are the principal and the most effective class of drugs to reduce serum cholesterol levels. Statins have been also shown to have anti-thrombotic effects in addition to lower cholesterol and to reduce cardiovascular events, including myocardial infarction, stroke, and death in patients with or without coronary artery disease symptoms. The actions of these drugs extend far beyond cholesterol reduction and involve non-lipid-related mechanisms that modify endothelial functions, immunoinflammatory responses, smooth muscle cell activation, proliferation and migration, atherosclerotic plaque stability and thrombus formation. Thus statins may offer an interesting prophylactic therapeutic approach for APS without the risks associated with anticoagulation. The aim of this study was to evaluate whether statins could modify the APLA-induced adhesion molecule expression by endothelial cells.