C. Introduction
Chronic congestive heart failure represents a major cause of cardiovascular morbidity and mortality in developed countries. It is caused by the loss of functional heart muscle, which is due either to ischemic heart disease or the presence of dysfunctional muscle resulting from a variety of causes, including hypertension, viruses, and idiopathic factors. Following myocardial infarction for example, functional contracting cardiomyocytes are replaced with nonfunctional scar tissue. This ventricular remodeling leads to ventricle dilatation and progressive heart failure which constitute a major clinical problem (Grounds et al., 2002). The remodeling process is characterized by the removal of necrotic cardiomyocytes accompanied by granulation tissue formation with the simultaneous induction of neovascularization in the peri-infarcted bed. The latter is a prerequisite for the survival of surrounding hypertrophied but viable cardiomyocytes and the prevention of further cardiomyocyte loss by apoptosis.
Several treatments for coronary artery disease are available, which reduce the symptoms and improve the quality of life of CHF patients. They include medical treatments (anticoagulants, b-blocker, angiotensin-converting-enzyme inhibitors, etc), effective percutaneous and surgical revascularization and cardiac pacing systems. Cardiac transplantation remains, however, the ultimate solution for end-stage heart failure. However, the shortage of donor hearts, the complications of immunosuppression, the failure of grafted organs and, not at last, the advanced age of patients suffering from CHF limit the utility of cardiac transplantation significantly.
Cell therapy as a mean to repair damaged tissues unable to heal is an increasingly attractive concept in modern transplantation medicine. For many clinical situations, i.e. congestive heart failure, Parkinson's disease, diabetes, traumatic injuries (spinal cord) and iatrogenic destruction of the cell (chemotherapy), replacement of lost cells would be the ideal treatment. In many cases, however, the development of cell treatment approaches is hampered by an increasing lack of donors or by the lack of cells suitable for transplantation.
C.1. Seeking stem cells desperately
Which cells could be used to repair a failing heart and restore its contractile power?
Two major types of stem cells would eventually fulfill suitable characteristics: 1) embryonic stem cells (ESC), derived from the inner cell mass of the blastocyst, and 2) adult somatic stem cells isolated form several organs (bone marrow, skeletal muscle, brain, etc).
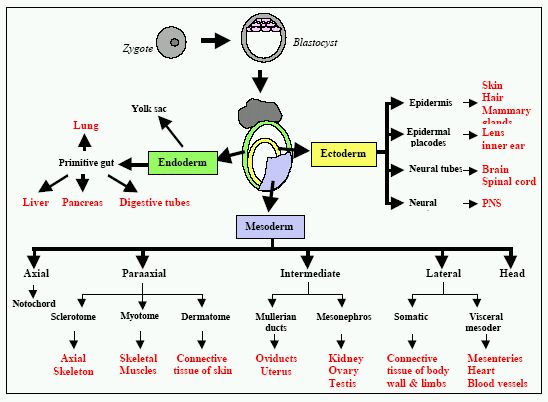
What is a stem cell? Stem cells are commonly defined as undifferentiated cells. They have the ability to differentiate into virtually all kinds of cell types, a capacity that becomes progressively restricted with development. As shown in Figure I, they have two important characteristics that distinguish them from other types of cells. First, as unspecialized cells, they can proliferate and renew themselves for long periods through cell division. The second is that under certain physiologic or experimental conditions, they can be induced to become cells with special functions. As the matter of fact, they provide a theoretically inexhaustible supply of cells that, depending on type can give rise to some or all body tissues.

Stem cells are typically found in the embryo and fetus. In the adult body, they have been identified in various tissue niches, including bone marrow, brain, liver, and skin, as well as in the circulation. They have been termed "adult stem cells". An extremely attractive concept is that adult stem cells could be harvested from a patient, induced to specialize in culture, then incorporated into a tissue construct, and put back into the same individual when repair become necessary, bypassing the need for immunosuppression.
The experimental donor cells for cellular cardiomyoplasty (CCM), as an alternative treatment of cardiovascular disease, include skeletal myoblasts, bone marrow-derived Mesenchymal stem cells, purified (enriched) haematopoietic stem cell populations and blood and bone marrow-derived endothelial progenitor cells.
C.2. Embryonic stem cells (ESC)
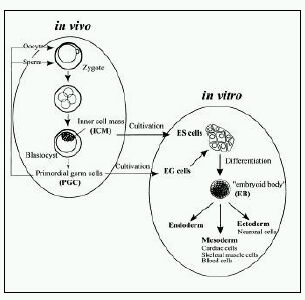
Pluripotent murine stem cells are derived from two main embryonic sources: ESC from the blastocyst and EG cells from the gonadal ridge of the embryo after gastrulation (Figure II). The successful derivation of murine ESC from the inner cell mass of mouse blastocytes was achieved in 1981 (Martin, 1981), while embryonic germ (EG) cells have been isolated and cultured from primordial germ cells (Stewart et al., 1994).

These cells were shown to be pluripotent, i.e., capable of forming all mature cell phenotypes derived from the three embryonic layers: endoderm, ectoderm, and mesoderm, as shown in Figure III.
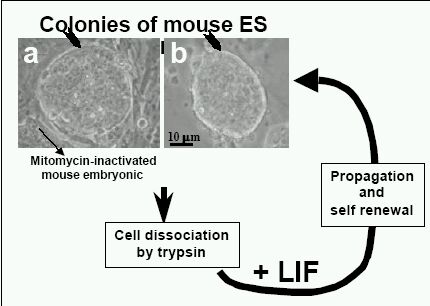
In vitro, murine ESC remain undifferentiated when grown in the presence of leukemia inhibitory factor (LIF) and for some cell lines, cultured on murine embryonic fibroblast (MEF) as feeder cells. The mechanism to maintain stem cell as a undifferentiated state is linked to the LIF/Stat3 signaling pathway and the transcription factor Oct-3/4, but it is still unclear how these components works together (Niwa, 2001). When LIF or feeder cells are withdrawn, most types of ESC differentiate spontaneously to form aggregates called embryoid bodies (EBs). Embryoid bodies are comprised of the heterogeneous cells that derived from all three germ layers. These tri-dimensional cell-cell contacts allow the formation of heterogeneous cultures of differentiated cell types including cardiomyocytes, hematopoietic cells, endothelial cells, neurons, skeletal muscle chondrocytes, adipocytes, liver, and pancreatic islets.
When murine ESC were cultured into embryoid bodies, some differentiated into a diverse range of cardiomyocyte phenotypes, including ventricular, atrial, sinus nodal, Purkinje, and pacemaker-like cells. These different cardiac phenotypes exhibit developmentally controlled expression of cardiac-specific genes, structural proteins, ion channels, and receptors, and can be distinguished on the basis of their action potentials. A few cell areas on the embryoid bodies display spontaneous contractile activity under light microscope, and are identifiable as ESC-derived cardiomyocytes. Since ESC are pluripotent, considering their use raises the potential risk of teratoma formation. Thus, the purification of differentiated ESC-derived cardiomyocytes from cultures is a key issue. Unfortunately, so far, none of the approaches used on murine ESC can give 100% yield of cells with the required phenotype.
C.3. No stem cells in the heart?
In the case of the heart, no local stem cells have been so far identified. Cardiomyocytes undergo terminal differentiation soon after birth and are generally considered to irreversibly withdrawn from the cell cycle. Cardiomyocyte DNA synthesis occurs primarily in uteri, with proliferating cells decreasing from 33% at mid-gestation to 2% at birth(MacLellan and Schneider, 2000). Therefore, upon injury, the adult heart results incapable to regenerate the damaged tissue which instead become fibrotic.
On note is the fact that the group of Piero Anversa was the only one to report that, in humans, some ventricular cardiomyocytes may have the capacity to proliferate or at least to undergo nuclear replication in response to ischemic injury. The dividing myocytes have been identified on the basis of immunohistochemical staining of proliferating nuclear structures such as Ki67 and cell surface expression of specific surface markers CD117. (Itescu et al., 2003). However, this is still a matter of debate and convincing proofs of cell division as a general event are still pending. It remains to be determined where does the dividing cells which homed to the damaged myocardium come from? Are those a resident source of cardiac stem cells, or do they come from a renewable source of circulating bone marrow-derived stem cells? It is not very clear so far (Anversa et al., 2003; Beltrami et al., 2001).
C.4. Cell therapy of the heart
The cell-based myocardial repair technology "cellular cardiomyoplasty" (CCM), attempt to regenerate functioning muscle in previously infarcted, scarred or dysfunctional myocardial tissue after transplantation of myogenic cells. The use of such a cell therapy approach to replace lost cardiomyocytes with new engraftable ones would represent an invaluable, low-invasiveness technique for treatment of heart failure as an alternative to whole heart transplantation. Replacement and regeneration of functional cardiac muscle after ischemia could be achieved either by stimulating proliferation of endogenous mature cardiomyocytes or by implanting exogenous donor-derived or allogeneic cardiomyocytes. The newly formed cardiomyocytes must integrate precisely into the existing myocardial wall to augment contractile function of the residual myocardium in a synchronized manner and avoid alteration in the electrical condition and syncytial contraction of the heart (Itescu et al., 2003).
To date, many types of cells have been tested as a source of cell therapy for the augmentation of myocardial performance in different experimental models of heart failure. Those include fetal cardiomyocytes (Leor et al., 1996; Li et al., 1996; Reinecke et al., 1999), ESC-derived cardiomyocytes (Min et al., 2002), skeletal myoblasts(Murry et al., 1996; Taylor et al., 1998), immortalized myoblasts(Robinson et al., 1996), fibroblasts(Hutcheson et al., 2000), smooth muscle cells (Li et al., 1999), fibroblasts (Murry et al., 1996), adult cardiac-derived cells(Li et al., 2000), and bone marrow-derived stem cells (Orlic et al., 2001). Different heart models have been used to study the effect of cell engraftment in the heart, e.g. normal heart tissue, cryoinjuried heart tissue, ischemic heart, infarcted scar tissue, dilated cardiomyopathy heart.
C.4.1. ESC into the heart
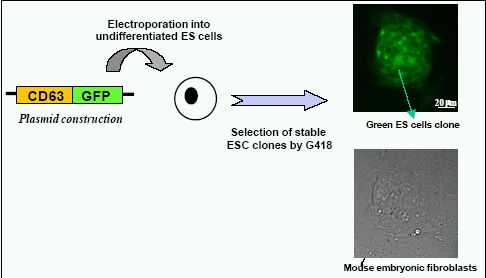
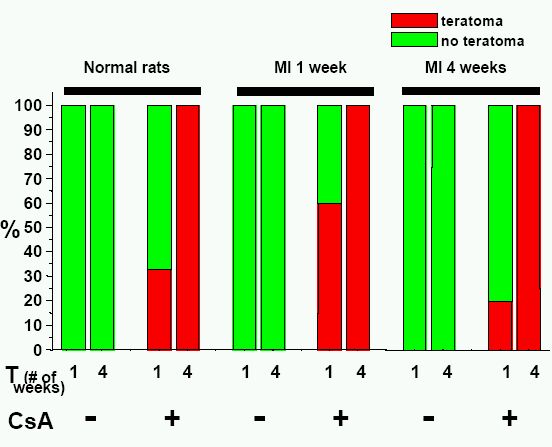
Several groups have demonstrated the in vivo feasibility of the intra-cardiac implantation of ESC. Klug et al. were the first to engraft genetically-modified and differentiated murine ESC-derived cardiomyocytes into the left ventricular free wall of mdx mice(Klug et al., 1996). Pure cardiomyocyte cultures were obtained by stable transfecting ESC with a transgene comprised of the a-cardiac myosin heavy chain (MHC) promoter driving a neomycin resistance gene. This construct being expressed only in cardiac cells, in the presence of neomycin (G418) it allowed the only survival of ESC-derived cardiomyocytes. The successful engraftment of donor ESC was confirmed by immunopositivity for dystrophin, and engrafted cells were found to be aligned with the host cardiomyocytes. More recently, Min et al. implanted cardiomyocytes derived from the D3-ESC line into a rat model of ischemic heart. Cardiomyocytes were selected from embryoid bodies by dissecting the spontaneously beating clusters via a sterile micropipette. After transfection with a green fluorescent protein (GFP) marker to identify survival of engrafted ESC, transplantation was performed within 30 minutes after induction of MI, created by ligation of the left anterior descending coronary artery. Under these conditions, the cardiac function was significantly improved 6 weeks after cell transplantation in MI animals compared with the MI control group (Min et al., 2002). However, some areas with undifferentiated cells were still observed, indicating that their extraction from the EBs was poor. Recently, Behfar et al reported that undifferentiated ESC directly implanted into an infarcted heart seems able to commit to the cardiac phenotype. Their study suggested that ESC differentiation require a paracrine pathway in the heart. In vitro, pretreatment of embryonic stem cells with TGFb growth factor members results in embryoid bodies with greater areas of cardiac differentiation (increased beating areas) and normal sarcomeric organization (as revealed by immunostaining). 5 weeks after the In vivo transplantation of undifferentiated ESC carrying the ECFP (cyan fluorescent protein) marker under the control of the cardiac a-actin promoter, they identified ESC-derived blue cardiomyocytes by immunostaining. The left ventricular ejection fraction (measured by echocardiography) was improved in a small number animals (n=3), as compared with control group (Behfar et al., 2002). More recently, Johkura et al. transplanted ESC-derived cardiomyocytes into the retroperitoneum of the adult nude mice. These myocytes proliferated, differentiated and remained viable and contractile for up to 30 days in the ectopic site around large blood vessels. However, contamination of the donor cells with the residual ESC committed to other lineages was likely to occur, even if the embryoid body outgrowths were transplanted after beating cardiomyocytes had appeared. This leaded to the formation of teratoma in the host retroperitoneum (Johkura et al., 2003).
In summary, the main issues which limit the research and the use of ESC for CCM include difficulties in obtaining pure and sufficient number of ES-derived cardiomyocytes, especially of ventricular- like cardiomyocytes, high risk of teratoma formation and immune rejection.
C.4.2. Human ESC
Presently, several human ESC lines are available since their first isolation by Thomson in 1998 (Thomson et al., 1998). A registry of them is published by the NIH web site (http://stemcells.nih.gov/registry/). The usage of human ESC as a resource for cell therapy is presently an intensive field of research (Mummery et al., 2003; Nir et al., 2003). At a basic research level, many biological differences should exist between the murine and human ESC and further fundamental studies are required before to investigate the suitability and feasibility of using human ES-derived cardiomyocytes for cell transplantation in humans. From a legal and ethical point of view, research involving human embryonic cells is highly controversial and many countries are reviewing their legislation. Importantly, main ethical issues are raised concerning the derivation of human ESC from human in vitro fertilized embryos, the moral status of the embryo, and the acceptability of using such derived cells for therapeutic purposes. The application of human ESC therapy for the treatment of cardiac diseases in humans is far from being a reality.
C.4.3. Skeletal muscle cells into the heart
The growth and repair of skeletal muscle is usually initiated by the activation of a population of muscle precursors, called satellite cells. Satellite cells normally lie near the basal lamina of the skeletal muscle fibers and can differentiated into myofibers. Following tissue injury, they are rapidly mobilized, proliferate and fuse, thereby effecting repair and regeneration of the damaged fibers.
Cardiac and skeletal muscles have a similar sophisticated organization of contractile proteins into sarcomeres and are collectively referred to as striated muscles. Growth of the heart is generally characterized by division of muscle cells during the embryonic stages of life, followed after birth by into a post-mitotic state. The postnatal capacity for cell replication during growth and regeneration of cardiac and skeletal muscle cells is markedly different. Skeletal muscle cells are multinucleated and can readily regenerate from precursor cells (satellite cells / myoblasts), whereas post-natal cardiac muscle is incapable of tissue repair, since cardiac cells exit the cell cycle soon after birth and become post-mitotic (either in vivo or in vitro).
Autologous skeletal myoblasts appear to be the most well studied and best first generation cells for cardiac repair. This process was pioneered in the 1980s (Sola et al., 1985), and has been applied clinically with varied success. The whole procedure includes extraction of the myoblasts from skeletal muscle, expansion in tissue culture, and injection into the heart muscle. For the success of cell transplantation, the introduced donor cells must be able to survive in their host environment. Intramuscular injection of cultured isolated myoblasts in classical myoblast transfer therapy shows that there is a massive and rapid necrosis of donor myoblasts, with 90% dead within the first hour after injection (Beauchamp et al., 1999). Host natural killer cells appear to play a particularly important role in this rapid death of cultured donor myoblasts.
In vivo, in various species, such as rabbits and rats, engraftment of skeletal myoblasts were shown colonized injured cardiac tissue(Murry et al., 1996; Pouzet et al., 2000; Taylor et al., 1998). A survival up to 12 weeks after transplantation was observed in normal heart (Reinecke et al., 2002), and up to 18 weeks in cryoinjured myocardium(Reffelmann and Kloner, 2003). An important factor is that skeletal myoblasts are relatively resistant to ischemia and, in contrast to cardiomyocytes (witch injure rapidly within 20 min), they can withstand several hours of severe ischemia without becoming irreversibly injured. Several studies reported improvement of left ventricular performance after myoblast transplantation, e.g. reducing left ventricular dilatation, increasing ex vivo systolic pressures and improving in vivo exercise capacity (Atkins et al., 1999; Jain et al., 2001).
However, the outcome of engraftment of skeletal myoblasts still remains highly controversial. Some reports claim that transplanted skeletal myoblasts could differentiate and develop into striated cells within the damaged myocardium, thus preventing the progressive ventricular dilatation by improving heart function (Taylor, 2001). On the other hand, some investigators have reported negative results and adverse effects. The systematic investigations by Reinecke et al. did not support the concept of a true transdifferentiation into cardiomyocytes as demonstrated by the lack of a-myosin-heavy-chain, cardiac troponin I, or atrial natriuretic peptide- expression in the grafts. Murry et al. also concluded that skeletal satellite muscle cells differentiate into mature skeletal muscle and do not express cardiac-specific genes after grafting into the heart, lacking any transdifferentiation potential (Reinecke et al., 2002).
Recently, attempts to use autologous skeletal satellite cells to repair damaged hearts have received considerable attention for the possibility of autotransplantation into human myocardium. The autologous origin would clearly overcomes all problems related to availability, ethics and immunogenicity, key factors for large-scale clinical applicability (Menasche, 2003). Menasche et al. first reported a three-steps protocol in a CHF patient in 2001 (Hagege et al., 2003; Menasche et al., 2003). A muscular biopsy was retrieved from the thigh under local anesthesia and, after enzymatic and mechanical dissociation, the cells were grown for 2-3 weeks in culture, providing at least 400x106cells, 50% of which were myoblasts. These cells are then reimplanted across the post-infarcted scar at the time of coronary bypass graft surgery (Menasche, 2003).
Several clinical trials are presently running in several countries, with different preliminary results. Some trials reported the presence of arrhythmia and sudden death, which leading to the suggestion to implant AICD´s on the time of procedure. The need of randomized trials is claimed in many conferences and meetings.
There are many important questions remain to be answered for the clinical use of myoblasts. They are related to cell survival, integration, differentiation, and functional effect. E.g. for the long-term benefit of skeletal myoblasts transplantation myocardial injury, cells must be able to survive for years in the heart. The long-term fate of myoblasts is unclear. The effect of transplanted myoblasts on electrical stability of the heart, as they do not form normal electrical junctions with the host, it will increase the risk of arrhythmia. In a phase I human clinical trial, 4 out of 10 cases showed sustained ventricular tachycardia during the early (3 weeks) postoperative period (Menasche et al., 2003). The mechanisms of these arrhythmias might be inhomogeneity in action potential conduction creating reentry pathways. So, how myoblasts electrically integrate into the surrounding myocardium is still a big issue from the clinical point of view. Can myoblasts function as cardiac-like myocytes and thus improve contraction, including adaptation to chronic workload and integration into the host, or can myoblasts just prevent further deterioration of the injured heart? How do we control continuing proliferation after transplantation to avoid undesirable disturbance of local left ventricular geometry? All those questions still need to be answered.
C.4.4. Bone Marrow stem cells
Bone marrow is a mesodermal-derived tissue and contains haematopoietic stem cells (HSCs) and mesenchymal stem cells (MSCs), which may both, be derived from a common primitive blast-like cell. The HSC are archetypal stem cells. They have the ability to balance self-renewal against differentiation cell fate decisions They are multipotent, a single stem cell producing at least eight to ten distinct lineages of mature cells. HSC have an extensive proliferative capacity that yields a large number of mature progeny. These cells are rare, comprising only 1/10'000 to 100'000 of total blood cells, and can be obtained from the bone marrow, peripheral blood umbilical cord and fetal liver(Bonnet, 2002; Preston et al., 2003). Krause et al., demonstrated that a single HSC was not only able to repopulate the haematopoietic system in irradiated mice, but also differentiated into lung epithelium, skin, liver and the gastrointestinal tract (Krause et al., 2001).
Figure V illustrates the transdifferentiation potential observed either in culture or after in vivo injection of cells.
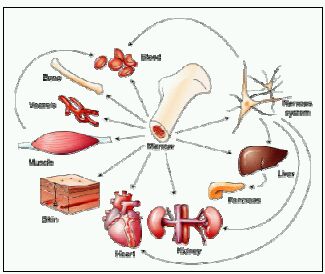
Too good to be true? Studies in mice have yielded evidence, now being reassessed, that stem cells from the bone marrow compartment can produce progeny in different organs. Bone marrow, which has several types of stem cells, seems particularly versatile.
MSCs have the main capacity to differentiate, both in vivo and in vitro, into osteoblast, chondroblasts, and adipocytes when exposed to the appropriate stimuli. Approximately 30% of human marrow aspirate cells adhering to plastic are considered to be MSCs. They are in general more difficult to characterized than the HSC populations. MSCs have been functionally identified in adult murine and human bone marrow by their ability to differentiate to lineages of diverse mesenchymal tissues. Those include bone, cartilage, fat, tendon, and both skeletal and cardiac muscle, which express specific surface markers but lack of haematopoietic lineage markers such as CD34 or CD 45. Adult mouse MSCs in culture were reported to generate spontaneously beating cardiomyocytes(Makino et al., 1999). When MSCs are pre-exposed to 5-azacytidine they are capable of differentiating into skeletal and cardiac muscle phenotypes (Makino et al., 1999). However the reproducibility of this treatment is highly questioned.
Recently, a number of studies have shown that bone marrow stem cells transplantation is beneficial for myocardial repair/regeneration in different animals and human being. Kamihata et al. injected autologous bone marrow-derived mononuclear cells into myocardial infarcted zone of swine(Kamihata et al., 2001). Three weeks later, regional blood flow and capillary densities were significantly higher, and cardiac function was improved. They concluded that bone marrow implantation may achieve optimal therapeutic angiogenesis through potent angiogenic ligands and cytokines secreted by those cells incorporated into foci of neovascularization. Orlic et al. reported that the injection into female mice of a subgroup of Lin-c-kit+ bone marrow cells from EGFP transgenic male donors at the borderline of an ischemic area resulted in the colonization of the infarcted area. Within 9 days, male EGFP-positive cells had proliferated in situ and expressed protein characteristic of cardiac tissue, including connexin43, thus suggesting intercellular communication (Orlic et al., 2001).
In human, several trials are ongoing. Perin and Dohmann recently reported that transendocardial injections of autologous mononuclear bone marrow cells in patients with end-stage ischemic heart disease could safely promote neovascularization and improve perfusion and myocardial contractility. 21 patients, all of them with previous myocardial infarction and documented with multivessel disease, were enrolled sequentially, with the first 14 patients assigned to the treatment group and the last 7 patients to the control group. Approximately 4 hours before the cell injection procedure, bone marrow (50ml) was aspirated; mononuclear cells were isolated by density gradient. The electromechanical map was used to identify viable myocardium for treatment; the cells were injected by NOGA catheter in the treatment group. Both of group patients underwent 2-month noninvasive follow-up, and treated patients along underwent a 4-month invasive follow-up. At 2-months, there was a significant reduction in total reversible defect and improvement in global left ventricular function within the two groups on quantitative SPECT analysis. At 4 months, the ejection fraction improved from a baseline of 20% to 29% (p=0.003), concomitantly to a reduction in end-systolic volume (p=0.03) in the treated patients. The limitation of the study includes the small number of patients, the short period of follow-up, and no placebo as control (Perin et al., 2003). Tse et al demonstrated somewhat similar results (Tse et al., 2003). They studied percutaneous delivery (via the Biosense Electromechanical NOGA mapping catheter) of autologous bone-marrow-derived mononuclear cells in eight patients with stable angina. After 3 months of follow-up, there was improvement in myocardial perfusion, regional myocardial wall motion and thickening, but LVEF remained unchanged (Tse et al., 2003). Hamano and Stamm reported two studies with 5 and 6 patients study respectively(Hamano et al., 2001; Stamm et al., 2003), in which both of groups received autologous bone marrow cells at the time of coronary artery bypass grafting, and were followed up for 3 to 12 months. Their results showed an improvement of perfusion of the infarcted myocardium possibly due to neoangiogenesis.
Presently, bone marrow stem cells provide an interesting and promising option to restore myocardial viability. The transplantation in human study appears feasible, relatively safe and effective, no tumor formation has been scored. However, a major criticism to these preliminary trials is the fact that the number of patients is too small to derive a meaningful efficacy and definitive safety data. Also, no data are available about cell survival following intra-myocardial needle injection, or whether implanted cells did survive and differentiate along the cardiac myocyte and/or endothelial lineage. In addition, some studies in big animals reported that injected cells are very low in number and tend to disappear with time. Further studies are required to better characterize the phenotype and the fate of injected cells.
Moreover, the in vitro proliferation potential of MSCs is not yet reproducible in order to obtain a suitable number of differentiated and characterized cells. Clearly, cell duplication and reproduction is the first condition to fully recolonize a diseased myocardium and thus improve ventricular function. Another factor is if undifferentiated stem cells are used, a risk is still present of developing other type of tissue(Laham and Oettgen, 2003; Scorsin and Souza, 2001).
The following three tables (Table I, II, III) are taken from a review by L. Field (Dowell et al., 2003) and represent to date an extensive list of all the cell therapy studies published so far.



References in tables I-III:
1. R. SoRelle, Myoblast transplant to heart attempted. Circulation 102 (2000), pp.E9030-9031.
2. B.E. Strauer, M. Brehm, T. Zeus, M. Kostering, A. Hernandez, R.V. Sorg et al., Repair of infarcted myocardium by autologous intracoronary mononuclear bone marrow cell transplantation in humans. Circulation 106 (2002), pp. 1913-1918.
3. M.H. Soonpaa, G.Y. Koh, M.G. Klug and L.J. Field, Formation of nascent intercalated disks between grafted fetal cardiomyocytes and host myocardium. Science 264 (1994), pp. 98-101.
4. G.Y. Koh, M.H. Soonpaa, M.G. Klug, H.P. Pride, B.J. Cooper, D.P. Zipes et al., Stable fetal cardiomyocyte grafts in the hearts of dystrophic mice and dogs. J Clin Invest 96 (1995), pp. 2034-2042.
5. C.H. Van Meter, Jr., W.C. Claycomb, J.B. Delcarpio, D.M. Smith, H. deGruiter, F. Smart et al., Myoblast transplantation in the porcine model: a potential technique for myocardial repair. J Thorac Cardiovasc Surg 110 (1995), pp. 1442-1448.
6. J. Leor, M. Patterson, M.J. Quinones, L.H. Kedes and R.A. Kloner, Transplantation of fetal myocardial tissue into the infarcted myocardium of rat. A potential method for repair of infarcted myocardium?. Circulation 94 (1996), pp. II332-336.
7. R.K. Li, Z.Q. Jia, R.D. Weisel, D.A. Mickle, J. Zhang, M.K. Mohabeer et al., Cardiomyocyte transplantation improves heart function. Ann Thorac Surg 62 (1996), pp. 654-660 discussion pp. 660-661 .
8. A.L. Connold, R. Frischknecht and G. Vrbova, A simple method for cardiac surgery in rats. Cell Transplant 5 (1996), pp. 405-409.
9. S. Gojo, S. Kitamura, W.T. Germeraad, Y. Yoshida, K. Niwaya and K. Kawachi, Ex vivo gene transfer into myocardium using replication-defective retrovirus. Cell Transplant 5 (1996), pp. S81-84.
10. A.L. Connold, R. Frischknecht, M. Dimitrakos and G. Vrbova, The survival of embryonic cardiomyocytes transplanted into damaged host rat myocardium. J Muscle Res Cell Motil 18 (1997), pp. 63-70.
11. M. Aoki, R. Morishita, J. Higaki, A. Moriguchi, S. Hayashi, H. Matsushita et al., Survival of grafts of genetically modified cardiac myocytes transfected with FITC-labeled oligodeoxynucleotides and the -galactosidase gene in the noninfarcted area, but not the myocardial infarcted area. Gene Ther 4 (1997), pp. 120-127.
12. S. Gojo, S. Kitamura, O. Hatano, A. Takakusu, K. Hashimoto, Y. Kanegae et al., Transplantation of genetically marked cardiac muscle cells. J Thorac Cardiovasc Surg 113 (1997), pp. 10-18.
13. M. Scorsin, A.A. Hagege, F. Marotte, N. Mirochnik, H. Copin, M. Barnoux et al., Does transplantation of cardiomyocytes improve function of infarcted myocardium?. Circulation 96 (1997), pp. II188-193.
14. R.K. Li, D.A. Mickle, R.D. Weisel, M.K. Mohabeer, J. Zhang, V. Rao et al., Natural history of fetal rat cardiomyocytes transplanted into adult rat myocardial scar tissue. Circulation 96 (1997), pp. II179-186 discussion 186-187 .
15. Z.Q. Jia, D.A. Mickle, R.D. Weisel, M.K. Mohabeer, F. Merante, V. Rao et al., Transplanted cardiomyocytes survive in scar tissue and improve heart function. Transplant Proc 29 (1997), pp. 2093-2094.
16. M. Scorsin, A.A. Hagege, I. Dolizy, F. Marotte, N. Mirochnik, H. Copin et al., Can cellular transplantation improve function in doxorubicin-induced heart failure?. Circulation 98 (1998), pp. II151-155 discussion II155-156.
17. E. Watanabe, D.M. Smith, Jr., J.B. Delcarpio, J. Sun, F.W. Smart, C.H. Van Meter, Jr. et al., Cardiomyocyte transplantation in a porcine myocardial infarction model. Cell Transplant 7 (1998), pp. 239-246.
18. T. Sakai, R.K. Li, R.D. Weisel, D.A. Mickle, Z.Q. Jia, S. Tomita et al., Fetal cell transplantation: a comparison of three cell types. J Thorac Cardiovasc Surg 118 (1999), pp. 715-724.
19. H. Reinecke, M. Zhang, T. Bartosek and C.E. Murry, Survival, integration, and differentiation of cardiomyocyte grafts: a study in normal and injured rat hearts. Circulation 100 (1999), pp. 193-202.
20. R.K. Li, Z.Q. Jia, R.D. Weisel, D.A. Mickle, A. Choi and T.M. Yau, Survival and function of bioengineered cardiac grafts. Circulation 100 (1999), pp. II63-69.
21. J. Leor, S. Aboulafia-Etzion, A. Dar, L. Shapiro, I.M. Barbash, A. Battler et al., Bioengineered cardiac grafts: a new approach to repair the infarcted myocardium?. Circulation 102 (2000), pp. III56-61.
22. M. Scorsin, A. Hagege, J.T. Vilquin, M. Fiszman, F. Marotte, J.L. Samuel et al., Comparison of the effects of fetal cardiomyocyte and skeletal myoblast transplantation on postinfarction left ventricular function. J Thorac Cardiovasc Surg 119 (2000), pp. 1169-1175.
23. T.S. Li, K. Hamano, K. Kajiwara, M. Nishida, N. Zempo and K. Esato, Prolonged survival of xenograft fetal cardiomyocytes by adenovirus-mediated CTLA4-Ig expression. Transplantation 72 (2001), pp. 1983-1985.
24. S. Etzion, A. Battler, I.M. Barbash, E. Cagnano, P. Zarin, Y. Granot et al., Influence of embryonic cardiomyocyte transplantation on the progression of heart failure in a rat model of extensive myocardial infarction. J Mol Cell Cardiol 33 (2001), pp. 1321-1330.
25. H. Yokomuro, R.K. Li, D.A. Mickle, R.D. Weisel, S. Verma and T.M. Yau, Transplantation of cryopreserved cardiomyocytes. J Thorac Cardiovasc Surg 121 (2001), pp. 98-107.
26. Y. Sakakibara, K. Tambara, F. Lu, T. Nishina, G. Sakaguchi, N. Nagaya et al., Combined procedure of surgical repair and cell transplantation for left ventricular aneurysm: an experimental study. Circulation 106 (2002), pp. I193-197.
27. Y. Sakakibara, K. Tambara, F. Lu, T. Nishina, N. Nagaya, K. Nishimura et al., Cardiomyocyte transplantation does not reverse cardiac remodeling in rats with chronic myocardial infarction. Ann Thorac Surg 74 (2002), pp. 25-30.
28. Y. Sakakibara, K. Nishimura, K. Tambara, M. Yamamoto, F. Lu, Y. Tabata et al., Prevascularization with gelatin microspheres containing basic fibroblast growth factor enhances the benefits of cardiomyocyte transplantation. J Thorac Cardiovasc Surg 124 (2002), pp. 50-56.
29. A. Ruhparwar, J. Tebbenjohanns, M. Niehaus, M. Mengel, T. Irtel, T. Kofidis et al., Transplanted fetal cardiomyocytes as cardiac pacemaker. Eur J Cardiothorac Surg 21 (2002), pp. 853-857.
30. W. Roell, Z.J. Lu, W. Bloch, S. Siedner, K. Tiemann, Y. Xia et al., Cellular cardiomyoplasty improves survival after myocardial injury. Circulation 105 (2002), pp. 2435-2441.
31. Rubart M, Soonpaa MH, Nakajima H, Nakajima H, Pasumarthi K, Field LJ. In-situ 2-photon microscopy reveals functional integration of grafted fetal cardiomyocytes in the adult host myocardium. American Heart Association 2002 Scientific Sessions, Abstract #104755.
32. M. Scorsin, F. Marotte, A. Sabri, O. Le Dref, M. Demirag, J.L. Samuel et al., Can grafted cardiomyocytes colonize peri-infarct myocardial areas?. Circulation 94 (1996), pp. II337-340.
33. T. Matsushita, M. Oyamada, H. Kurata, S. Masuda, A. Takahashi, T. Emmoto et al., Formation of cell junctions between grafted and host cardiomyocytes at the border zone of rat myocardial infarction. Circulation 100 (1999), pp. II262-268.
34. T. Imanishi, C.E. Murry, H. Reinecke, T. Hano, I. Nishio, W.C. Liles et al., Cellular FLIP is expressed in cardiomyocytes and down-regulated in TUNEL-positive grafted cardiac tissues. Cardiovasc Res 48 (2000), pp. 101-110.
35. M. Zhang, D. Methot, V. Poppa, Y. Fujio, K. Walsh and C.E. Murry, Cardiomyocyte grafting for cardiac repair: graft cell death and anti-death strategies. J Mol Cell Cardiol 33 (2001), pp. 907-921.
36. S. Miyagawa, Y. Sawa, S. Taketani, N. Kawaguchi, T. Nakamura, N. Matsuura et al., Myocardial regeneration therapy for heart failure: hepatocyte growth factor enhances the effect of cellular cardiomyoplasty. Circulation 105 (2002), pp. 2556-2561.
37. J. Muller-Ehmsen, K.L. Peterson, L. Kedes, P. Whittaker, J.S. Dow, T.I. Long et al., Rebuilding a damaged heart: long-term survival of transplanted neonatal rat cardiomyocytes after myocardial infarction and effect on cardiac function. Circulation 105 (2002), pp. 1720-1726.
38. J. Muller-Ehmsen, P. Whittaker, R.A. Kloner, J.S. Dow, T. Sakoda, T.I. Long et al., Survival and development of neonatal rat cardiomyocytes transplanted into adult myocardium. J Mol Cell Cardiol 34 (2002), pp. 107-116.
39. T. Shimizu, M. Yamato, Y. Isoi, T. Akutsu, T. Setomaru, K. Abe et al., Fabrication of pulsatile cardiac tissue grafts using a novel 3-dimensional cell sheet manipulation technique and temperature-responsive cell culture surfaces. Circ Res 90 (2002), p. e40.
40. T. Eschenhagen, M. Didie, J. Heubach, U. Ravens and W.H. Zimmermann, Cardiac tissue engineering. Transplant Immunol 9 (2002), pp. 315-321.
41. T. Sakai, R.K. Li, R.D. Weisel, D.A. Mickle, E.J. Kim, S. Tmita et al., Autologous heart cell transplantation improves cardiac function after myocardial injury. Ann Thorac Surg 68 (1999), pp. 2074-2080 discussion pp. 2080-2081 .
42. K.J. Yoo, R.K. Li, R.D. Weisel, D.A. Mickle, Z.Q. Jia, E.J. Kim et al., Heart cell transplantation improves heart function in dilated cardiomyopathic hamsters. Circulation 102 (2000), pp. III204-209.
43. R.K. Li, R.D. Weisel, D.A. Mickle, Z.Q. Jia, E.J. Kim, T. Sakai et al., Autologous porcine heart cell transplantation improved heart function after a myocardial infarction. J Thorac Cardiovasc Surg 119 (2000), pp. 62-68.
44. T.M. Yau, K. Fung, R.D. Weisel, T. Fujii, D.A. Mickle and R.K. Li, Enhanced myocardial angiogenesis by gene transfer with transplanted cells. Circulation 104 (2001), pp. I218-222.
45. G.Y. Koh, M.H. Soonpaa, M.G. Klug and L.J. Field, Long-term survival of AT-1 cardiomyocyte grafts in syngeneic myocardium. Am J Physiol 264 (1993), pp. H1727-1733.
46. J.B. Delcarpio and W.C. Claycomb, Cardiomyocyte transfer into the mammalian heart. Cell-to-cell interactions in vivo and in vitro. Ann NY Acad Sci 752 (1995), pp. 267-285.
47. G.Y. Koh, M.G. Klug, M.H. Soonpaa and L.J. Field, Differentiation and long-term survival of C2C12 myoblast grafts in heart. J Clin Invest 92 (1993), pp. 1548-1554.
48. G.Y. Koh, S.J. Kim, M.G. Klug, K. Park, M.H. Soonpaa and L.J. Field, Targeted expression of transforming growth factor-beta 1 in intracardiac grafts promotes vascular endothelial cell DNA synthesis. J Clin Invest 95 (1995), pp. 114-121.
49. S.W. Robinson, P.W. Cho, H.I. Levitsky, J.L. Olson, R.H. Hruban, M.A. Acker et al., Arterial delivery of genetically labelled skeletal myoblasts to the murine heart: long-term survival and phenotypic modification of implanted myoblasts. Cell Transplant 5 (1996), pp. 77-91.
50. H. Reinecke and C.E. Murry, Transmural replacement of myocardium after skeletal myoblast grafting into the heart. Too much of a good thing?. Cardiovasc Pathol 9 (2000), pp. 337-344.
51. K. Suzuki, N.J. Brand, R.T. Smolenski, J. Jayakumar, B. Murtuza and M.H. Yacoub, Development of a novel method for cell transplantation through the coronary artery. Circulation 102 (2000), pp. III359-364.
52. K. Suzuki, R.T. Smolenski, J. Jayakumar, B. Murtuza, N.J. Brand and M.H. Yacoub, Heat shock treatment enhances graft cell survival in skeletal myoblast transplantation to the heart. Circulation 102 (2000), pp. III216-221.
53. C.E. Murry, R.W. Wiseman, S.M. Schwartz and S.D. Hauschka, Skeletal myoblast transplantation for repair of myocardial necrosis. J Clin Invest 98 (1996), pp. 2512-2523.
54. H. Reinecke, G.H. MacDonald, S.D. Hauschka and C.E. Murry, Electromechanical coupling between skeletal and cardiac muscle. Implications for infarct repair. J Cell Biol 149 (2000), pp. 731-740
55. M. Jain, H. DerSimonian, D.A. Brenner, S. Ngoy, P. Teller, A.S. Edge et al., Cell therapy attenuates deleterious ventricular remodeling and improves cardiac performance after myocardial infarction. Circulation 103 (2001), pp. 1920-1927.
56. D. Marelli, C. Desrosiers, M. el-Alfy, R.L. Kao and R.C. Chiu, Cell transplantation for myocardial repair: an experimental approach. Cell Transplant 1 (1992), pp. 383-390.
57. D. Marelli, F. Ma and R.C. Chiu, Satellite cell implantation for neomyocardial regeneration. Transplant Proc 24 (1992), p. 2995.
58. A. Zibaitis, D. Greentree, F. Ma, D. Marelli, M. Duong and R.C. Chiu, Myocardial regeneration with satellite cell implantation. Transplant Proc 26 (1994), p. 3294.
59. R.C. Chiu, A. Zibaitis and R.L. Kao, Cellular cardiomyoplasty: myocardial regeneration with satellite cell implantation. Ann Thorac Surg 60 (1995), pp. 12-18.
60. P.D. Yoon, R.L. Kao and G.J. Magovern, Myocardial regeneration. Transplanting satellite cells into damaged myocardium. Tex Heart Inst J 22 (1995), pp. 119-125.
61. D.A. Taylor, S.C. Silvestry, S.P. Bishop, B.H. Annex, R.E. Lilly, D.D. Glower et al., Delivery of primary autologous skeletal myoblasts into rabbit heart by coronary infusion: a potential approach to myocardial repair. Proc Assoc Am Phys 109 (1997), pp. 245-253.
62. D.A. Taylor, B.Z. Atkins, P. Hungspreugs, T.R. Jones, M.C. Reedy, K.A. Hutcheson et al., Regenerating functional myocardium: improved performance after skeletal myoblast transplantation. Nat Med 4 (1998), pp. 929-933
63. J. Dorfman, M. Duong, A. Zibaitis, M.P. Pelletier, D. Shum-Tim, C. Li et al., Myocardial tissue engineering with autologous myoblast implantation. J Thorac Cardiovasc Surg 116 (1998), pp. 744-751.
64. B.Z. Atkins, M.T. Hueman, J. Meuchel, K.A. Hutcheson, D.D. Glower and D.A. Taylor, Cellular cardiomyoplasty improves diastolic properties of injured heart. J Surg Res 85 (1999), pp. 234-242.
65. B.Z. Atkins, M.T. Hueman, J.M. Meuchel, M.J. Cottman, K.A. Hutcheson and D.A. Taylor, Myogenic cell transplantation improves in vivo regional performance in infarcted rabbit myocardium. J Heart Lung Transplant 18 (1999), pp. 1173-1180.
66. B.Z. Atkins, C.W. Lewis, W.E. Kraus, K.A. Hutcheson, D.D. Glower and D.A. Taylor, Intracardiac transplantation of skeletal myoblasts yields two populations of striated cells in situ. Ann Thorac Surg 67 (1999), pp. 124-129.
67. K.A. Hutcheson, B.Z. Atkins, M.T. Hueman, M.B. Hopkins, D.D. Glower and D.A. Taylor, Comparison of benefits on myocardial performance of cellular cardiomyoplasty with skeletal myoblasts and fibroblasts. Cell Transplant 9 (2000), pp. 359-368.
68. R.J. Lee, M.L. Springer, W.E. Blanco-Bose, R. Shaw, P.C. Ursell and H.M. Blau, VEGF gene delivery to myocardium: deleterious effects of unregulated expression. Circulation 102 (2000), pp. 898-901.
69. K. Suzuki, B. Murtuza, R.T. Smolenski, I.A. Sammut, N. Suzuki, Y. Kaneda et al., Cell transplantation for the treatment of acute myocardial infarction using vascular endothelial growth factor-expressing skeletal myoblasts. Circulation 104 (2001), pp. I207-212.
70. C. Rajnoch, J.C. Chachques, A. Berrebi, P. Bruneval, M.O. Benoit and A. Carpentier, Cellular therapy reverses myocardial dysfunction. J Thorac Cardiovasc Surg 121 (2001), pp. 871-878.
71. B. Pouzet, J.T. Vilquin, A.A. Hagege, M. Scorsin, E. Messas, M. Fiszman et al., Factors affecting functional outcome after autologous skeletal myoblast transplantation. Ann Thorac Surg 71 (2001), pp. 844-850 discussion pp. 850-851 .
72. E.G. Chedrawy, J.S. Wang, D.M. Nguyen, D. Shum-Tim and R.C. Chiu, Incorporation and integration of implanted myogenic and stem cells into native myocardial fibers: anatomic basis for functional improvements. J Thorac Cardiovasc Surg 124 (2002), pp. 584-590.
73. N. Dib, E.B. Diethrich, A. Campbell, N. Goodwin, B. Robinson, J. Gilbert et al., Endoventricular transplantation of allogenic skeletal myoblasts in a porcine model of myocardial infarction. J Endovasc Ther 9 (2002), pp. 313-319
74. S. Etzion, I.M. Barbash, M.S. Feinberg, P. Zarin, L. Miller, E. Guetta et al., Cellular cardiomyoplasty of cardiac fibroblasts by adenoviral delivery of MyoD ex vivo: an unlimited source of cells for myocardial repair. Circulation 106 (2002), pp. I125-130.
75. K. Suzuki, B. Murtuza, L. Heslop, J.E. Morgan, R.T. Smolenski, N. Suzuki et al., Single fibers of skeletal muscle as a novel graft for cell transplantation to the heart. J Thorac Cardiovasc Surg 123 (2002), pp. 984-992.
76. S. Ghostine, C. Carrion, L.C. Souza, P. Richard, P. Bruneval, J.T. Vilquin et al., Long-term efficacy of myoblast transplantation on regional structure and function after myocardial infarction. Circulation 106 (2002), pp. I131-136.
77. H. Reinecke, V. Poppa and C.E. Murry, Skeletal muscle stem cells do not transdifferentiate into cardiomyocytes after cardiac grafting. J Mol Cell Cardiol 34 (2002), pp. 241-249.
78. M.G. Klug, M.H. Soonpaa, G.Y. Koh and L.J. Field, Genetically selected cardiomyocytes from differentiating embryonic stem cells form stable intracardiac grafts. J Clin Invest 98 (1996), pp. 216-224.
79. A. Behfar, L.V. Zingman, D.M. Hodgson, J.M. Rauzier, G.C. Kane, A. Terzic et al., Stem cell differentiation requires a paracrine pathway in the heart. FASEB J 16 (2002), pp. 1558-1566.
80. J.Y. Min, Y. Yang, K.L. Converso, L. Liu, Q. Huang, J.P. Morgan et al., Transplantation of embryonic stem cells improves cardiac function in postinfarcted rats. J Appl Physiol 92 (2002), pp. 288-296.
81. Y. Yang, J.Y. Min, J.S. Rana, Q. Ke, J. Cai, Y. Chen et al., VEGF enhances functional improvement of postinfarcted hearts by transplantation of ESC-differentiated cells. J Appl Physiol 93 (2002), pp. 1140-1151.
82. S. Tomita, R.K. Li, R.D. Weisel, D.A. Mickle, E.J. Kim, T. Sakai et al., Autologous transplantation of bone marrow cells improves damaged heart function. Circulation 100 (1999), pp. II247-256.
83. J.S. Wang, D. Shum-Tim, J. Galipeau, E. Chedrawy, N. Eliopoulos and R.C. Chiu, Marrow stromal cells for cellular cardiomyoplasty: feasibility and potential clinical advantages. J Thorac Cardiovasc Surg 120 (2000), pp. 999-1005.
84. K.W. Liechty, T.C. MacKenzie, A.F. Shaaban, A. Radu, A.M. Moseley, R. Deans et al., Human mesenchymal stem cells engraft and demonstrate site-specific differentiation after in utero transplantation in sheep. Nat Med 6 (2000), pp. 1282-1286
85. K.A. Jackson, S.M. Majka, H. Wang, J. Pocius, C.J. Hartley, M.W. Majesky et al., Regeneration of ischemic cardiac muscle and vascular endothelium by adult stem cells. J Clin Invest 107 (2001), pp. 1395-1402.
86. C.E. Murry, M. Rubart, M. Soonpaa, H. Nakajima, H. Nakajima and L.J. Field, Absence of cardiac differentiation in hematopoietic stem cells transplanted into normal and injured hearts. Circulation (abstract) 104 (2001), p. II599.
87. J.S. Wang, D. Shum-Tim, E. Chedrawy and R.C. Chiu, The coronary delivery of marrow stromal cells for myocardial regeneration: pathophysiologic and therapeutic implications. J Thorac Cardiovasc Surg 122 (2001), pp. 699-705.
88. B.E. Strauer, M. Brehm, T. Zeus, N. Gattermann, A. Hernandez, R.V. Sorg et al., (Intracoronary, human autologous stem cell transplantation for myocardial regeneration following myocardial infarction). Dtsch Med Wochenschr 126 (2001), pp. 932-938.
89. D. Orlic, J. Kajstura, S. Chimenti, I. Jakoniuk, S.M. Anderson, B. Li et al., Bone marrow cells regenerate infarcted myocardium. Nature 410 (2001), pp. 701-705.
90. K. Hamano, T.S. Li, T. Kobayashi, K. Hirata, M. Yano, M. Kohno et al., Therapeutic angiogenesis induced by local autologous bone marrow cell implantation. Ann Thorac Surg 73 (2002), pp. 1210-1215.
91. Y. Jiang, B.N. Jahagirdar, R.L. Reinhardt, R.E. Schwartz, C.D. Keene, X.R. Ortiz-Gonzalez et al., Pluripotency of mesenchymal stem cells derived from adult marrow. Nature 418 (2002), pp. 41-49.
92. T.C. Mackenzie, A.F. Shaaban, A. Radu and A.W. Flake, Engraftment of bone marrow and fetal liver cells after in utero transplantation in MDX mice. J Pediatr Surg 37 (2002), pp. 1058-1064.
93. T. Saito, J.Q. Kuang, B. Bittira, A. Al-Khaldi and R.C. Chiu, Xenotransplant cardiac chimera: immune tolerance of adult stem cells. Ann Thorac Surg 74 (2002), pp. 19-24 discussion p. 24 .
94. S. Tomita, D.A. Mickle, R.D. Weisel, Z.Q. Jia, L.C. Tumiati, Y. Allidina et al., Improved heart function with myogenesis and angiogenesis after autologous porcine bone marrow stromal cell transplantation. J Thorac Cardiovasc Surg 123 (2002), pp. 1132-1140. .
95. C. Toma, M.F. Pittenger, K.S. Cahill, B.J. Byrne and P.D. Kessler, Human mesenchymal stem cells differentiate to a cardiomyocyte phenotype in the adult murine heart. Circulation 105 (2002), pp. 93-98.
96. A.J. Wagers, R.I. Sherwood, J.L. Christensen and I.L. Weissman, Little evidence for developmental plasticity of adult hematopoietic stem cells. Science 297 (2002), pp. 2256-2259.
97. R.E. Bittner, C. Schofer, K. Weipoltshammer, S. Ivanova, B. Streubel, E. Hauser et al., Recruitment of bone-marrow-derived cells by skeletal and cardiac muscle in adult dystrophic mdx mice. Anat Embryol (Berlin) 199 (1999), pp. 391-396.
98. D.L. Clarke, C.B. Johansson, J. Wilbertz, B. Veress, E. Nilsson, H. Karlstrom et al., Generalized potential of adult neural stem cells. Science 288 (2000), pp. 1660-1663.
99. G. Condorelli, U. Borello, L. De Angelis, M. Latronico, D. Sirabella, M. Coletta et al., Cardiomyocytes induce endothelial cells to trans-differentiate into cardiac muscle: implications for myocardium regeneration. Proc Natl Acad Sci USA 98 (2001), pp. 10733-10738.