

« Back to all publications
Download this list in a RIS file or a BIB file or a PDF file
|
||||||||
Femtosecond transient absorption anisotropy studies have been performed for two porphycenes of different symmetry. In 2,7,12,17-tetraphenylporphycene, the chemical identity of two trans forms implies a change in the S0âS1 transition-moment direction upon tautomerization. Exploiting this phenomenon, the rates of double hydrogen transfer in both the S0 and S1 states (1.4Ã1012 sâ1 and 2.7Ã1011 sâ1, respectively) have been determined by performing time-resolved anisotropy studies. In the asymmetric 9-amino-2,7,12,17-tetraphenylporphycene, tautomerization occurs with a similar rate in the ground state. In the S1 state, the reaction is hindered in its vibrationally relaxed form, but the excitation spectra suggest that it may occur for an unrelaxed molecule. Unlike all porphycenes that have been studied so far, 9-amino-2,7,12,17-tetraphenylporphycene does not reveal significant changes in anisotropy owing to intramolecular double hydrogen transfer; rather, the transition-moment directions are similar in the two tautomeric forms. Analysis of the molecular orbitals allows for an explanation of the âlockingâ of the transition moments: it is due to a large splitting of the two HOMO orbitals, which retain the order of their energies in the two tautomers. | ||||||||



|
 |
|||||||
This study addresses the free energy dependence of charge recombination following photoinduced bimolecular electron transfer in three different solvents of either inert (acetonitrile and benzyl acetate) or reactive (N,N-dimethylaniline) character. Femtosecond time-resolved fluorescence and transient absorption have been used to determine the time scales for charge recombination. In pure N,N-dimethylaniline, charge recombination is found to be substantially slower than charge separation in a range of driving forces covering 1.5 eV. In all three solvents, the so-called Marcus inverted region is clearly observed for charge recombination. Additionally, the charge recombination step is found to be influenced by the solvent relaxation dynamics. A diffusion-reaction equation approach using an electron transfer model accounting for solvent relaxation is used to rationalize the experimental results. | ||||||||
 |
|
|||||||
Norrish-type-II reaction on a semiquinone radical: Stable semiquinone radicals serve as novel molecular platforms on which a Norrish-type-II photoreaction can be initiated. A detailed reaction scheme involving a 1,5-hydrogen transfer followed by a cyclization step that finally leads to a new CâC bond formation could be verified. Transient absorption spectroscopy and DFT calculations trace convincingly the intermediates and transition states along the reaction path (see scheme). | ||||||||
|
 |
|||||||
Liquid/liquid interfaces play a crucial role in numerous areas of science. However, direct spectroscopic access to this thin (â¼1 nm) region is not possible with conventional optical methods. After a brief review of the most used techniques to perform interfacial optical spectroscopy, we will focus on time-resolved surface second harmonic generation, which allows the measurement of the excited-state dynamics of probe molecules at interfaces. By comparing these dynamics with those measured in bulk solutions, precious information on the properties of the interfacial region can be obtained. To illustrate this, several studies performed in our group will be presented. | ||||||||
 |
|
|||||||
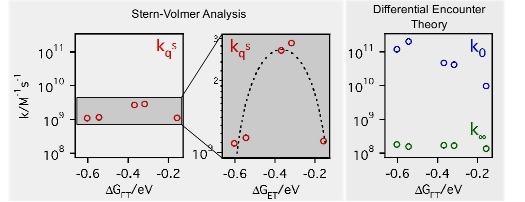
The effect of viscosity on the bimolecular electron transfer quenching of a series of coumarins by N,N-dimethylaniline was investigated using steady-state and time-resolved fluorescence spectroscopy. The data reveal that the static and transient stages of the quenching become dominant as viscosity increases. When extracting the quenching rate constants using a simple SternâVolmer analysis, a decrease of the rate constant with increasing driving force is observed above ~2 cP. However, this apparent Marcus inverted region, already reported several times with the same system in micelles and room temperature ionic liquids, totally vanishes when analyzing the data with a model accounting for the static and transient stages of the quenching. It appears that the apparent Marcus inverted region arises from the neglect of these quenching regimes together with the use of fluorophores with different excited-state lifetimes. | ||||||||
|
 |
|||||||
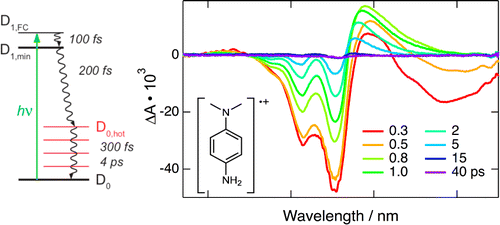
The excited-state dynamics of a series of Wursterâs salts (p-phenylenediamine radical cations) with different subtituents on the nitrogen atoms was investigated under a variety of experimental conditions using a combination of ultrafast spectroscopic techniques. At room temperature, the lifetime of the lowest excited state of all radical cations is on the order of 200 fs, independently of the solvent, that is, water, nitriles, alcohols, and room-temperature ionic liquid. On the other hand, all cations, except that with the bulky nitrogen substituents, become fluorescent below 120 K. The observed dynamics can be accounted for by the presence of a conical intersection between the D1Â and D0Â states. For the cations with a small nitrogen substituent, this conical intersection could be accessed through a twist of one amino group, as already suggested for Wursterâs Blue. However, this coordinate cannot be invoked for the cation with bulky nitrogen subtituents, and more probably, pyramidalization of the nitrogen center and/or deformation of the phenyl ring play an important role. Consequently, the excited-state dynamics of these structurally very similar Wursterâs salts involves different decay mechanisms. | ||||||||
 |
|
|||||||
Although liquid/liquid and air/liquid interfaces are omnipresent, very little is known up to now about the dynamics of processes occurring at such interfaces. As a detailed understanding of these processes could be of invaluable technological, environmental, and medical importance, considerable effort has been invested over the last two decades in developing new interface-selective techniques that allow for gaining further insight into the dynamics of these processes. Whereas several major results have been achieved that helped to contribute to a deeper understanding, there are still many aspects concerning the properties of liquid interfaces that are not yet fully understood. In this Perspective, the work that has been carried out so far on photoinduced interfacial dynamics will be reviewed and the current challenges in this still emerging field of research discussed. | ||||||||
|
 |
|||||||
Molecular systems where several apparently equivalent charge separation pathways exist upon photoexcitation are presented. They encompass MQn (nâ¥2) architectures, where M is a chromophore and Q an electron transfer quencher (either donor or acceptor), and MâM systems where M acts as both electron donor and acceptor. In all cases, charge separation involves symmetry breaking. The conditions for such process to be operative as well as the origin of the symmetry breaking are discussed. | ||||||||
 |
|
|||||||
The photophysical properties of a series of helicene cations in various solvents have been investigated using stationary and time-resolved spectroscopy. These compounds fluoresce in the near infrared region with a quantum yield ranging between 2 and 20% and a lifetime between 1 and 12 ns, depending of the solvent. No clear solvent dependence could be recognized except for a decrease of fluorescence quantum yield and lifetime with increasing hydrogen-bond donating ability of the solvent. In water, the helicene cations undergo aggregation. This effect manifests itself by the presence of a slow fluorescence decay component, whose amplitude increases with dye concentration, and by a much slower decay of the polarization anisotropy in water compared to an organic solvent of similar viscosity. However, aggregation has essentially no effect on the stationary fluorescence spectrum, whereas relatively small changes can be seen in the absorption spectrum. Analysis of the dependence of aggregation on the dye concentration reveals that the aggregates are mostly dimers and that the aggregation constant is substantially larger for hetero- than homochiral dimers. | ||||||||
|
 |
|||||||
The fluorescence quenching of 3-cyanoperylene upon electron transfer from N,N-dimethylaniline in three room-temperature ionic liquids (RTILs) and in binary solvent mixtures of identical viscosity has been investigated using steady-state and time-resolved fluorescence spectroscopy. This study was stimulated by previous reports of bimolecular electron transfer reactions faster by one or several orders of magnitude in RTILs than in conventional polar solvents. These conclusions were usually based on a comparison with data obtained in low-viscous organic solvents and extrapolated to higher viscosities and not by performing experiments at similar viscosities as those of the RTILs, which we show to be essential. Our results reveal that (i) the diffusive motion of solutes in both types of solvents is comparable, (ii) the intrinsic electron transfer step is controlled by the solvent dynamics in both cases, being slower in the RTILs than in the conventional organic solvent of similar viscosity, and (iii) the previously reported reaction rates much larger than the diffusion limit at low quencher concentration in RTILs originate from a neglect of the static and transient stages of the quenching, which are dominant in solvents as viscous as RTILs. | ||||||||
 |
|
|||||||
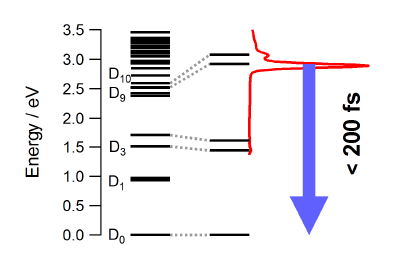
The photophysical properties of the free neutral radical galvinoxyl were studied by a combination of femtosecond time-resolved spectroscopy and quantum chemical calculations. The electronic absorption spectrum is dominated by an intense band at 430 nm that is ascribed to the D9,10âD0 transitions. Upon photoexcitation at 400 nm, the population of the D9,10 states decays within less than 200 fs to the electronic ground state. This ultrafast internal conversion does not involve intramolecular modes with large amplitude motion as the measured dynamics does not show any significant dependence on the environment, but is most probably facilitated by a high density of electronic states of different character. Depending on the solvent, a weak transient band due to the galvinoxylate anion is also observed. This closed-shell species, which is fluorescent although its deactivation is also dominated by non-radiative decay, is generated upon biphotonic ionization of the solvent and electron capture. The ultrashort excited-state lifetime of the galvinoxyl radical precludes photoinduced disproportionation previously claimed to be at the origin of the formation of both anion and cation. | ||||||||
Download this list in a RIS file or a BIB file or a PDF file
Contact:
Eric Vauthey
Physical Chemistry Department - Sciences II - University of Geneva
30, Quai Ernest Ansermet - CH-1211 Geneva 4 (Switzerland)
© All rights reserved by Eric Vauthey and the University of Geneva
Design and code by Guillaume Duvanel