

« Back to all publications
Download this list in a RIS file or a BIB file or a PDF file
|
 |
|||||||
The excited state and electron injection dynamics of three new organic sensitizers, comprising a triphenylamine moiety connected by an ethenylene (CâC double-bond) or ethynylene (CâC triple-bond) Ï-spacer to an electron-withdrawing benzothiazole bearing a cyanoacrylic acid anchoring group, have been studied using a combination of steady-state and femtosecond-resolved spectroscopies. The measurements were carried out for the three dyes in predominantly neutral and completely deprotonated forms in liquid solutions and bound on nanocrystalline TiO2 and Al2O3 thin films. In addition, quantum-chemical calculations were performed to predict absorption spectra of the sensitizers and their corresponding cation radicals. Time-resolved fluorescence (TRF) measurements on TiO2 indicate that electron injection takes place on a <0.2 ps time scale. Transient electronic absorption (TA) measurements provide evidence for the formation of radical cations not only in dye-sensitized TiO2 films but also in Al2O3 ones. The cation lifetime in Al2O3 is significantly shorter compared to TiO2, indicating a faster recombination of injected electrons with the dye cations. In addition, the ground-state bleach band in dye-sensitized TiO2 films experiences a gradual red-shift, which is indicative of a transient Stark effect. Finally, femtosecond transient absorption measurements in the IR region point to an ultrafast generation of injected electrons for all dyes. A faster recombination of the injected electrons with the dye cations is observed for the sensitizer decorated with auxiliary electron-donating methoxy groups on the triphenylamine moiety. | ||||||||



 |
|
|||||||
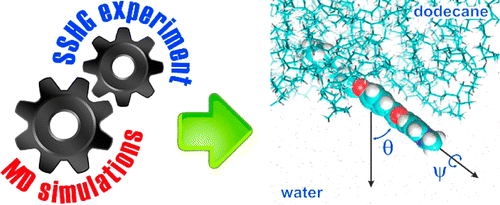
The second-order nonlinear response of two dyes adsorbed at the dodecane/water interface was investigated by surface second harmonic generation (SSHG). These dyes consist of the same chromophoric unit, 2-pyridinyl-5-phenyloxazole, with an alkyl chain located at the two opposite ends. The analysis of the polarization dependence of the SSHG intensity as usually performed points to similar tilt angles of the two dyes with respect to the interface but does not give information on the absolute direction. Molecular dynamics (MD) simulations reveal that both dyes lie almost flat at the interface but have opposite orientations. A refined SSHG data analysis with the width of the orientational distribution yields tilt angles that are in very satisfactory agreement with the MD simulations. | ||||||||
|
 |
|||||||
Because of their key role in many areas of science and technology, bimolecular photoinduced electron transfer reactions have been intensively studied over the past five decades. Despite this, several important questions, such as the absence of the Marcus inverted region or the structure of the primary reaction product, have only recently been solved while others still remain unanswered. Ultrafast spectroscopy has proven to be extremely powerful to monitor the entire electron transfer process and to access, with the help of state-of-the-art theoretical models of diffusion-assisted reactions, crucial information like e.g. the intrinsic charge separation dynamics beyond the diffusion limit. Additionally, extension of these experimental techniques to other spectral regions than the UV-visible, such as the infrared, has given a totally new insight into the nature, the structure and the dynamics of the key reaction intermediates, like exciplexes and ions pairs. In this perspective, we highlight these recent progresses and discuss several aspects that still need to be addressed before a thorough understanding of these processes can be attained. | ||||||||
|
||||||||
Configurationally stable diaza[4]helicenes have been prepared in two steps by using a particularly facile NâN bond-cleavage reaction. The synthetic procedure uses hydrazine (NH2NH2) for the introduction of a single nitrogen atom. The strategy is general, modular and highly tolerant to functional groups. A mechanistic rationale is proposed for the spontaneous NâN bond-cleavage reaction. The resulting helical quinacridines are dyes that present absorption and emission properties that can be modulated as a function of pH; the pink quinacridine and green protonated forms (pKa â 9.0) display distinct optical features in the near-IR region. Single enantiomers were obtained by chiral stationary phase HPLC resolution. The absolute configurations were assigned by comparison of the ECD spectra of the conjugated acids with those of known dialkylquinacridinium derivatives. A rather high racemization barrier was measured by means of variable-temperature ECD experiments (ÎGâ¡ = 30.7â±â4.0 kcalâmolâ1 at 140 °C). | ||||||||
|
||||||||
A C-C bond forming reaction occurs spontaneously between tris-(2,6-dimethoxyphenyl)carbenium ion and indoles / anilines. The carbocation acts both as electrophile and oxidant. Effective cationic Ï-conjugated dyes are formed resulting in a strong hyper- and bathochromism | ||||||||
 |
|
|||||||
The excited-state dynamics of a diketopyrrolopyrrole (DPP) derivative with pushâpull substituents has been investigated in a variety of solvents and at the dodecane/water and dodecane/heavy-water interfaces using a combination of ultrafast spectroscopic techniques, including transient electronic absorption and time-resolved surface second-harmonic generation. Whereas the photophysics of a nonpolar DPP analogue is mostly independent of the solvent, the fluorescence decay of the pushâpull DPP accelerates strongly by going from aprotic to protic solvents. As this effect increases with the polarity and the hydrogen-bond-donating ability of the solvent, it is attributed to the occurrence of H-bond-assisted nonradiative deactivation induced by the charge-transfer character of the excited state that favors the coupling of the molecule to the H-bond network of the solvent. At the dodecane/water interface, the excited-state lifetime is longer by a factor of ca. 20 than that estimated in pure water and increases further by a factor of about 3 when going to the dodecane/heavy-water interface. This isotope effect, that is more than twice as strong as that measured in bulk solutions, and molecular dynamic simulations indicate that the slowing down of the dynamics at the interface cannot be solely ascribed to a reduced accessibility of the DPP molecule to the aqueous phase. The slower excited-state decay is rather assigned to the conjunction of several effects, such as the strengthening of the H-bond network formed by the interfacial water molecules and the lower local polarity of the interfacial region. | ||||||||
|
 |
|||||||
The dynamics of photoinduced electron transfer between polar acceptors and donors has been investigated in apolar solvents using femtosecond-resolved fluorescence spectroscopy. It was found to be ultrafast and to continuously accelerate by varying the excitation wavelength from the maximum to the red edge of the absorption band of the acceptor, the overall difference being as large as a factor 4â5. This violation of the KashaâVavilov rule is explained by a correlation between the composition of the acceptor environment and its transition energy, that is, the more donors around an acceptor, the longer its absorption wavelength, and the faster the quenching. Because of preferential solvation, this dependence is already observed at low quencher concentrations. This effect, which requires quenching to be faster than the fluctuations of the environment composition, should be quite general for photoinduced charge transfer processes in low-polarity, viscous, or rigid media, such as those used in organic optoelectronic devices. | ||||||||
 |
|
|||||||
The excited-state dynamics of rhodamine 6G (R6G) has been investigated in aqueous solution using ultrafast transient absorption spectroscopy and at the dodecane/water interface using the femtosecond time-resolved surface second harmonic generation (SSHG) technique. As the R6G concentration exceeds ca. 1 mM in bulk water, both R6G monomers and aggregates are excited to a different extent when using pump pulses at 500 and 530 nm. The excited-state lifetime of the monomers is shortened compared to dilute solutions because of the occurrence of excitation energy transfer to the aggregates, which themselves decay nonradiatively to the ground state with a ca. 70 ps time constant. At the dodecane/water interface, both monomers and aggregates contribute to the SSHG signal to an extent that depends on the bulk concentration, the pump and probe wavelengths, and the polarization of probe and signal beams. The excited-state lifetime of the monomers at the interface is of the order of a few picoseconds even at bulk concentrations where it is as large as several nanoseconds. This is explained by the relatively high interfacial affinity of R6G that leads to a large interfacial concentration, favoring aggregation and thus rapid excitation energy transfer from monomers to aggregates. | ||||||||
|
 |
|||||||
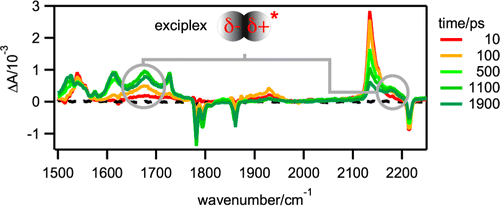
The dynamics of bimolecular photoinduced electron-transfer reactions has been investigated with three donor/acceptor (D/A) pairs in tetrahydrofuran (THF) and acetonitrile (ACN) using a combination of ultrafast spectroscopic techniques, including time-resolved infrared absorption. For the D/A pairs with the highest driving force of electron transfer, all transient spectroscopic features can be unambiguously assigned to the excited reactant and the ionic products. For the pair with the lowest driving force, three additional transient infrared bands, more intense in THF than in ACN, with a time dependence that differs from those of the other bands are observed. From their frequency and solvent dependence, these bands can be assigned to an exciplex. Moreover, polarization-resolved measurements point to a relatively well-defined mutual orientation of the constituents and to a slower reorientational time compared to those of the individual reactants. Thanks to the minimal overlap of the infrared signature of all transient species in THF, a detailed reaction scheme including the relevant kinetic and thermodynamic parameters could be deduced for this pair. This analysis reveals that the formation and recombination of the ion pair occur almost exclusively via the exciplex. | ||||||||
 |
|
|||||||
The excited-state dynamics of two multichromophoric arrays composed of a naphthalene diimide centre and four zinc or free-base porphyrins substituted on the naphthalene core via aniline bridges has been investigated using a combination of stationary and ultrafast spectroscopies. These pentads act as efficient antennae as they absorb over the whole visible region, with a band around 700 nm, associated with a transition to the S1 state delocalised over the whole arrays, and bands at higher energy due to transitions centred on the porphyrins. In non-polar solvents, population of these porphyrin states is followed by sub-picosecond internal conversion to the S1 state. The existence of a charge-separated state located above the S1 state could enhance this process. The decay of the S1 state is dominated by non-radiative deactivation on the 100 ps timescale, most probably favoured by the small S1-S0 energy gap and the very high density of vibrational states of these very large chromophores. In polar solvents, the charge-separated state lies just below the S1 state. It can be populated within a few picoseconds by a thermally-activated hole transfer from the S1 state as well as via sub-picosecond non-equilibrium electron transfer from vibrationally hot porphyrin excited states. Because of the small energy gap between the charge-separated state and the ground state, charge recombination is almost barrierless and occurs within a few picoseconds. Despite their very different driving forces, charge separation and recombination occur on similar timescales. This is explained by the electronic coupling that differs considerably for both processes. | ||||||||
|
 |
|||||||
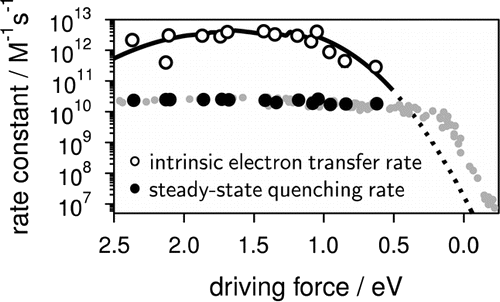
To access the intrinsic, diffusion free, rate constant of bimolecular photoinduced electron transfer reactions, fluorescence quenching experiments have been performed with 14 donor/acceptor pairs, covering a driving-force range going from 0.6 to 2.4 eV, using steady-state and femtosecond time-resolved emission, and applying a diffusion-reaction model that accounts for the static and transient stages of the quenching for the analysis. The intrinsic electron transfer rate constants are up to 2 orders of magnitude larger than the diffusion rate constant in acetonitrile. Above ~1.5 eV, a slight decrease of the rate constant is observed, pointing to a much weaker Marcus inverted region than those reported for other types of electron transfer reactions, such as charge recombination. Despite this, the driving force dependence can be rationalized in terms of Marcus theory. | ||||||||
Download this list in a RIS file or a BIB file or a PDF file
Contact:
Eric Vauthey
Physical Chemistry Department - Sciences II - University of Geneva
30, Quai Ernest Ansermet - CH-1211 Geneva 4 (Switzerland)
© All rights reserved by Eric Vauthey and the University of Geneva
Design and code by Guillaume Duvanel