|
|
-
Raman Optical Activity of (β)-citronellene
H. Hagemann, A. Lucken, D. Lovy and H. Bill
in "Proc. XIV th Conference on Raman Spectroscopy"
Eds. N.T. Yu, X.Y. Ku, J. Wiley and sons, Singapore,
14 (1994), p1072-1073


|
|
|
We present an ab initio molecular dynamics study of the structure and dynamics of a close-packed monolayer of Pb on the Ge(111) surface, with coverage FTHETA=4/3, at different temperatures. The room-temperature (â3 à â3 ) structure is characterized by large in-plane fluctuations of the Pb adatoms, and agrees well with recent x-ray standing wave data. At Tâ¼800 K we observe a (1Ã1) disordered structure showing in-plane diffusion of the Pb atoms. Disordering is confined to the plane of the overlayer. A strong correlation with the solid Ge substrate is present, leading to preferential residence sites and diffusion paths of the Pb atoms on the surface. The calculated local density of states for the high-temperature phase is found to agree with recent scanning tunnel microscope observations, which show a simply Pb-terminated (1Ã1) surface with an apparent coverageFTHETA=1. |
|
|
The absorption spectra of the ferrate (VI) ion (FeO2-4) in K2MO4 (M = S, Se, Cr, Mo) host lattices consist of a series of relatively weak bands at low energy, which can be assigned to transitions within the partially filled 3d shell and some intense bands at higher energy, which are assigned to ligand-to-metal charge transfer transitions (LMCT). In the near-infrared (NIR) region sharp lines are observed belonging to the spin-forbidden spin-flip transitions 3A2â 1E and 3A2  â 1A1. The lowest excited state is the 1E state, serving as initial state for 1E â 3A2 sharp-line luminescence at around 6200 cm-1. Another luminescence is observed centered at 9000 cm-1, which is assigned to the 3T2 â 3A2 transition. It is rather broad and three orders of magnitude weaker than the 1E luminescence at 30K as a result of efficient non-radiative relaxation processes to the 1E state. The temperature dependence of the total intensity and the lifetime of the 1E â 3A2 luminescence is understood within a complex scheme of radiative and non-radiative processes. |
|
|
A spectroscopic study of supersonic jetâcooled catechol (1,2âdihydroxybenzene) and its d1â and d2âisotopomers, deuterated at the hydroxy groups, was performed by resonant twoâphoton ionization (R2PI) and fluorescence emission techniques, and supplemented by molecularâbeam holeâburning experiments. The latter prove that one single rotamer of catechol is dominant under molecular beam conditions. The complicated vibrational structure in the S0âS1 spectrum from the 000 band to 400 cmâ1 above is not due to three different rotamers, as previously thought, but is due to the excitation of a vibrational progression associated mainly with the torsion of the hydroxy groups. The torsional bands are very prominent in the R2PI spectra, but are weak in the emission spectra. Detailed analysis of the torsional bands was based on a fit to the S1 and S0 state frequencies and the FranckâCondon factors in absorption and emission, using a doubleâminimum potential for the S1 state and a harmonic potential for the S0 state. In the S1 state one of the two âOâH torsional mode frequencies is lowered from Ï2â250 to â50 cmâ1, and the molecule is only quasiplanar with respect to the âOâH torsional coordinates. |
|
|
Mass-selective ground state vibrational spectroscopy of the jet-cooled carbazole·Ar complex was performed by populating ground-state levels via a pump/dump laser pulse sequence, followed by selective resonant two-photon ionization of the vibrationally relaxed complexes. Intra- and inter-molecular van der Waals modes in the S0 state are measurable with good signal/noise. The ground-state binding energy can be determined by detecting the negative signals resulting from loss of ground-state population via vibrational predissociation of the complex. |
|
|
The cyclic water trimer shows a fascinating complexity of its intermolecular potential-energy surface as a function of the three intermolecular torsional coordinates: there are six isometric but permutationally distinct minimum-energy structures of C1 symmetry, which can interconvert by torsional motions via six isometric transition states, also of C1 symmetry. A second type of interconversion can occur through different torsional motions via two C3 symmetric transition structures, and a third interconversion type via a planar C3h symmetric transition structure. The equivalence of the six minima is broken if the âfreeâ H atom of one H2O molecule in the cluster is chemically substituted, yielding three distinct conformers, which occur in enantiomeric pairs. Not all three conformers are necessarily locally stable minima; this depends on the substituent. The phenolâ(H2O)2, p-cyanophenolâ(H2O)2, 1-naphtholâ(H2O)2 and 2-naphtholâ(H2O)2 clusters, which are the phenyl, p-cyanophenyl and naphthyl derivatives of (H2O)3, were examined by resonant two-photon ionization spectroscopy in supersonic beams. These clusters exhibit S0â S1 vibronic spectra with very different characteristics. These reflect the number of cluster structures formed, their low-frequency intermolecular vibrations and indirectly give information about the cluster fluxionality. |
|
-
A polymeric two-dimensional mixed-metal network. Crystal structure and magnetic properties of {[P(Ph)4][MnCr(ox)3]}n
S. Decurtins, H.W. Schmalle, H.R. Oswald, A. Linden, J. Ensling, P. Gütlich and A. Hauser
Inorganica Chimica Acta, 216 (1-2) (1994), p65-73
Keywords: crystal structures, magnetism, manganese complexes, chromium complexes, dinuclear complexes
DOI:10.1016/0020-1693(93)03711-I | unige:3005 | Abstract | Article PDF
|
The mixed-metal ferromagnet {[P(Ph)4][MnCr(ox)3]}n, where Ph is phenyl and ox is oxalate, has been prepared and a two-dimensional network structure, extended by Mn(II)-ox-Cr(III) bridges, has been determined from single crystal X-ray data. Crystal data: space group R3c, a=b=18.783(3), c=57.283(24) Ã
, α=β=90, γ=120°, Z=24 (C30H20O12PCrMn). The magnetic susceptibility data obey the Curie-Weiss law in the temperature range 260â20 K with a positive Weiss constant of 10.5 K. The temperature dependence of the molar magnetization exhibits a magnetic phase transition at Tc=5.9 K. The structure is discussed in relation to the strategy for preparing molecular based ferromagnets and, in addition, it is a solution to the question of the dimensionality of the [MM'(ox)3]n network, which in principle can extend two- or three-dimensionally to the crystal lattice. The optical absorption spectra of the single crystals are assigned to the âCrO6' chromophores. Their polarization patterns reflect the electric dipole selection rules for D3 symmetry. A strong site selective luminescence from the chromium(III) 2E states is observed at low temperature and the system may be suitable for studying energy transfer mechanisms. |
|
|
| 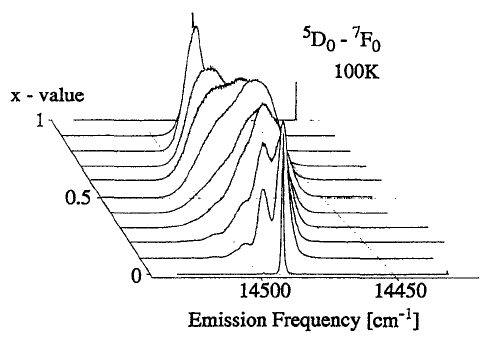 |
We have developed a model to describe the inhomogeneous broadening of optical spectra in the substitutionally disordered crystals. The comparison with the experimentalfâf fluorescence spectra of SrFClxBr1âx:Sm2+ (0â¤xâ¤1) allowed to establish, in a very detailed manner, the relationship between the inhomogeneous spectral distribution and the crystal structure around the Sm2+ impurity. |
|
|
Single crystals of the alkaline earth fluorides doped with silver were grown successfully. This paper presents details of the methodology. The as-grown crystals consist of colorless transparent and yellowish regions. The former were shown to contain the Ag+ ion and the latter also silver pairs, small clusters and probably colloidal aggregates. Complex optical absorption bands were observed in the samples of the former parts after they had been X-irradiated. The samples were subsequently exposed to extended series of physico-chemical treatments with the aim to obtain information regarding the electronic structures involved. The evolution was monitored with the aid of optical absorption experiments. Factor analysis technique is presented and was applied to uncover mutually independent contributions to these absorption spectra. We identified the Jahn-Teller Ag+ [Bill H. et al., Solid State Commun.70, 511 (1989)] and several centers which formally involve an Agâ ion. |
|
|
The spectroscopic properties of Ag+-doped strontium fluoride crystals were investigated at various temperatures, using absorption and fluorescence spectroscopies. The system exhibits a strong ultraviolet emission upon excitation into the two principal absorption bands. The azimuthal dependence of the degree of polarization of this luminescence is analysed, as well as its dynamics. The monovalent silver ions are shown to substitute for a host cation, with cubic symmetry. This is the first reported example of a cubic coordination for the Ag+ ions in an insulator. This cubic field, together with the strong ionic character of the framework, confers rather original spectroscopic properties to this system. The luminescence mechanisms are interpreted on the basis of the measured decay times and with the aid of energy diagram calculations. Two closed thermalized spin-orbit levels, with symmetry A2g and T2g respectively, are involved in the luminescence processes. The pure spin triplet A2g only emits at low temperatures (T<15 K), whereas the T2g level ( approximately 2% spin singlet character) emits in turn upon warming the crystal. One-dimensional configuration coordinate diagrams are proposed to interpret the peculiar temperature dependence of the emission band maximum. |
|
|
Transition metal chemistry contains a class of complex compounds for which the spin state of the central atom changes from high spin to low spin when the temperature is lowered. This is accompanied by changes of the magnetic and optical properties that make the thermally induced spin transition (also called spin crossover) easy to follow. The phenomenon is found in the solid state as well as in solution. Amongst this class, iron(II) spin crossover compounds are distinguished for their great variety of spin transition behavior; it can be anything from gradual to abrupt, stepwise, or with hysteresis effects. Many examples have been thoroughly studied by Mössbauer and optical spectroscopy, measurements of the magnetic susceptibilities and the heat capacities, as well as crystal structure analysis. Cooperative interactions between the complex molecules can be satisfactorily explained from changes in the elastic properties during the spin transition, that is, from changes in molecular structure and volume. Our investigations of iron(II) spin crossover compounds have shown that green light will switch the low spin state to the high spin state, which then can have a virtually unlimited lifetime at low temperatures (this phenomenom is termed light-induced excited spin state trapping - acronym: LIESST). Red light will switch the metastable high spin state back to the low spin state. We have elucidated the mechanism of the LIESST effect and studied the deactivation kinetics in detail. It is now well understood within the theoretical context of radiationless transitions. Applications of the LIESST effect in optical information technology can be envisaged. |
|
|
In der Ãbergangsmetallchemie gibt es eine Klasse von Komplexverbindungen, bei denen eine Temperaturerniedrigung einen Wechsel im Spinzustand des Zentralatoms vom High-Spin- in den Low-Spin-Zustand bewirkt. Dabei ändern sich die magnetischen und optischen Eigenschaften, über die der thermische Spinübergang (auch Spincrossover genannt) sehr gut verfolgt werden kann. Dieses Phänomen tritt sowohl in flüssiger Phase als auch im Festkörper auf. Eine herausragende Stellung nehmen Eisen(II) - Spincrossover - Verbindungen ein, in denen der Spinübergang im Festkörper auf sehr unterschiedliche Weise - graduell, abrupt, mit Hysterese oder stufenweise - verlaufen kann und mit MöÃbauer- und optischer Spektroskopie, mit magnetischen Suszeptibilitäts- und Wärmekapazitätsmessungen sowie durch Kristallstrukturanalyse intensiv untersucht worden ist. Die kooperative Wechselwirkung zwischen den einzelnen Komplexmolekülen kann befriedigend durch elastische Eigenschaften und durch die Ãnderung von Gestalt und Volumen der Komplexmoleküle beim Spinübergang erklärt werden. Bei Untersuchungen an Eisen(II)-Spincrossover-Verbindungen konnte man beobachten, daà sich der Low-Spin-Zustand mit grünem Licht in den High-Spin-Zustand umschalten läÃt, der bei tiefen Temperaturen eine nahezu unendlich lange Lebensdauer haben kann (LIESST = Light-Induced Excited Spin State Trapping). Mit rotem Licht läÃt sich der metastabile High-Spin- wieder in den Low-Spin-Zustand zurückschalten. Der Mechanismus des LIESST-Effekts ist aufgeklärt, die Zerfallskinetik im Detail untersucht und im Rahmen der Theorie strahlungsloser Ãbergänge verstanden. Anwendungen des LIESST-Effekts in der optischen Informationstechnik sind denkbar. |
|
|
A study of the effect of an external electric field on spectral holes burnt at different frequencies in the inhomogeneous absorption band of a centrosymmetric squaraine dye, bis [4-(diethylamino)-2-hydroxyphenyl] squaraine (DEAH), in polymers of different polarity is presented. Average matrix induced dipole moment differences of about 1 D and 0.37 D were measured in the directions parallel and perpendicular to the long axis of DEAH. In all polymers investigated, the induced dipole moment difference decreased from the higher to the lower frequencies. Solvatochromic shift measurements were performed in order to elucidate the origin of this effect. The matrix field inducing the dipole moment is also partially responsible for the frequency shift of the absorption of DEAH. With increasing matrix field, the absorptiion frequency is shifted to the blue due to electrostatic interaction with the local dipoles of DEAH. The contribution of the electrostatic interactions to the frequency shift is smaller than the dispersion interactions by two orders of magnitude in polystyrene, but increases slightly in more polar polymers. |
|
|
The free ion yield (R) resulting from the fluorescence quenching of 9,10-dicyanoanthracene (DCA) by various electron donors in acetonitrile has been studied using ns laser photoconductivity. The influence of the chemical nature of the doors is established in a general manner. For a given oxidation potential Eox(D/D+), the rate constant of geminate ion recombination, kbac, decreases significantly as the electronic delocalization of the donor increases. As a consequence multiple Marcus plots are observed in the inverted region. These plots show decreasing curvature when going from stilbenes to amines as donors. This fan effect is tentatively explained by considering the detailed roles of the parameters V, and h in the Marcus model. |
|
-
Picosecond Transient Grating Spectroscopy: An Application of Holography for Investigating Ultrafast Photoinduced Processes
E. Vauthey, Y. Pariat and A. Henseler
Chimia, 48 (9) (1994), p397-400
unige:3001 | Abstract | Article PDF
|
The principles of ps transient grating spectroscopy are presented. The capabilities of this technique for the study of photoinduced processes are illustrated by several new results. These include the observation of an anomalous effect in the reorientation dynamics of two ionic dyes in long-chain alkanenitriles, the measurement of the local viscosity in the interior of reverse micelles containing methanol, and the determination of the rate constant of separation of a geminate radical pair formed by photoinduced electron-transfer reaction. |
|
|
The rotational diffusion time of ruthenium (II)-bis(2,2'-bipyridine) (2,2'-biquinoleine) has been measured in polar solvents of different viscosity. The rotational dynamics can be explained in terms of the StokesâEinsteinâDebye hydrodynamics theory under stick boundary condition by considering the rotating body as a prolate spheroid enclosing the complex. This overestimates the intrinsic molecular volume by a factor of 1.5. The difference can be accounted for by solvent molecules intercalated in the interligand space and stabilized by electrostatic interaction with the charge of the metal atom. |
|
|
The rate of separation into free ions of a geminate ion pair generated by photoinduced electron transfer between 9,lO-anthraquinone excited to the lowest triplet state and 1,2,4-trimethoxybenzenein 1,1,2,2-tetrachloroethane has been measured at different temperatures by nanosecond time-resolved resonance Raman spectroscopy (TR3). The intrinsic activation energy for the separation amounts to 0.04 eV, suggesting that the center-to-center interionic distance in the geminate ion pair is about 7.5 A. The activation barrier is due to a loss of electrostatic stabilization upon separation to a distance of about 9.5 A, where a solvent molecule or part of it can interpenetrate to increase the solvation energy. This suggests that the geminate ion pair is a loose ion pair but is not truly solvent separated. |
|
|
The 3,6-substituted 1,2,4-trioxan-5-ones 11-14, on heating to 170-200°, underwent unimolecular thermolysis to generate electronically excited singlet ketones with an efficiency of ca. 0.2%. The chemiluminescence quantum yields (ΦoSCL) depended on the nature of the 6-substitutents and increased linearly with temperature. The Arrhenius activation energies were obtained by measuring the rate of decay of luminescence and determined as 22.9, 30.4, 35.6, and 34.2 kcal/mol for 11-14, respectively. Step analysis of the chemiluminescence of 14 afforded an average activation energy of 44.3 kcal/mol. This latter result is explicable in terms of two decomposition paths, higher and lower in energy, leading to excited and 'dark' products, respectively. The thermolysis of trioxanones 12-14 lacking a H-atom at the 6-position is interpreted as involving successive rupture of the peroxide bond, excision of ketone at the 3-substituted end, and loss of CO2, to finally produce ketone originating from the 6-position (see Scheme 4). |
|
|
Extensive ab initio calculations of the phenolâ
H2O complex were performed at the HartreeâFock level, using the 6â31G(d,p) and 6â311++G(d,p) basis sets. Fully energyâminimized geometries were obtained for (a) the equilibrium structure, which has a translinear H bond and the H2O plane orthogonal to the phenol plane, similar to (H2O)2; (b) the lowestâenergy transition state structure, which is nonplanar (C1 symmetry) and has the H2O moiety rotated by ±90°. The calculated MP2/6â311G++(d,p) binding energy including basis set superposition error corrections is 6.08 kcal/mol; the barrier for internal rotation around the H bond is only 0.4 kcal/mol. Intraâ and intermolecular harmonic vibrational frequencies were calculated for a number of different isotopomers of phenolâ
H2O. Anharmonic intermolecular vibrational frequencies were computed for several intermolecular vibrations; anharmonic corrections are very large for the β2 intermolecular wag. Furthermore, the H2O torsion Ï around the Hâbond axis, and the β2 mode are strongly anharmonically coupled, and a twoâdimensional Ï/β2 potential energy surface was explored. The role of tunneling splitting due to the torsional mode is discussed and tunnel splittings are estimated for the calculated range of barriers. The theoretical studies were complemented by a detailed spectroscopic study of hâphenolâ
H2O and dâphenolâ
D2O employing twoâcolor resonanceâtwoâphoton ionization and dispersed fluorescence emission techniques, which extends earlier spectroscopic studies of this system. The β1 and β2 wags of both isotopomers in the S0 and S1 electronic states are newly assigned, as well as several other weaker transitions. Tunneling splittings due to the torsional mode may be important in the S0 state in conjunction with the excitation of the intermolecular Ï and β2 modes. |
|
|
A combined experimental and theoretical study of the 2ânaphtholâ
H2O/D2O system was performed. Two different rotamers of 2ânaphthol (2âhydroxynaphthalene, 2HN) exist with the OâH bond in cisâ and transâposition relative to the naphthalene frame. Using HartreeâFock (HF) calculations with the 6â31G(d,p) basis set, fully energyâminimized geometries were computed for both cisâ and transâ2HNâ
H2O of (a) the equilibrium structures with transâlinear Hâbond arrangement and Cs symmetry and (b) the lowestâenergy transition states for H atom exchange on the H2O subunit, which have a nonplanar C1 symmetry. Both equilibrium and transition state structures are similar to the corresponding phenolâ
H2O geometries. The Hâbond stabilization energies with zero point energy corrections included are â5.7 kcal/mol for both rotamers, â2.3 kcal/mol stronger than for the water dimer, and correspond closely to the binding energy calculated for phenolâ
H2O at the same level of theory. Extension of the aromatic Ïâsystem therefore hardly affects the Hâbonding conditions. The barrier height to internal rotation around the Hâbond only amounts to 0.5 kcal/mol. Harmonic vibrational analysis was carried out at these stationary points on the HF/6â31G(d,p) potential energy surface with focus on the six intermolecular modes. The potential energy distributions and Mâmatrices reflect considerable mode scrambling for the deuterated isotopomers. For the aâ² intermolecular modes anharmonic corrections to the harmonic frequencies were evaluated. The β2 wag mode shows the largest anharmonic contributions. For the torsional mode Ï (H2O Hâatom exchange coordinate) the vibrational level structure in an appropriate periodic potential was calculated. On the experimental side resonantâtwoâphoton ionization and dispersed fluorescence emission spectra of 2HNâ
H2O and dâ2HNâ
D2O were measured. A detailed assignment of the bands in the intermolecular frequency range is given, based on the calculations. The predicted and measured vibrational frequencies are compared and differences discussed. |
|
|
The minimum energy structure of the cyclic water trimer, its stationary points, and rearrangement processes at energies <1 kcal/mol above the global minimum are examined by ab initio molecular orbital theory. Structures corresponding to stationary points are fully optimized at the HartreeâFock and secondâorder MøllerâPlesset levels, using the 6â311++G(d,p) basis; each stationary point is characterized by harmonic vibrational analyses. The lowest energy conformation has two free OâH bonds on one and the third OâH bond on the other side of an approximately equilateral hydrogenâbonded O...O...O (O3) triangle. The lowest energy rearrangement pathway corresponds to the flipping of one of the two free OâH bonds which are on the same side of the plane across this plane via a transition structure with this OâH bond almost within the O3 plane. Six distinguishable, but isometric transition structures of this type connect six isometric minimum energy structures along a cyclic vibrationalâtunneling path; neighboring minima correspond to enantiomers. The potential energy along this path has C6 symmetry and a very low barrier V6=0.1±0.1 kcal/mol. This implies nearly free pseudorotational interconversion of the six equilibrium structures. The corresponding anharmonic level structure was modeled using an internal rotation Hamiltonian. Two further lowâenergy saddle points on the surface are of second and third order; they correspond to crownâtype and planar geometries with C3 and C3h symmetries, respectively. Interconversion tunneling vibrations via these stationary points are also important for the water trimer dynamics. A unified and symmetryâadapted description of the intermolecular potential energy surface is given in terms of the three flipping coordinates of the OâH bonds. Implications of these results for the interpretation of spectroscopic data are discussed. |
|
|
The well-resolved absorption, excitation, and luminescence spectra of [Ir(ppy)2bpy]+ (ppyH = 2-phenylpyridine, bpy = 2,2'-bipyridine) in different media at cryogenic temperatures are presented. In solutions and glasses at ambient temperature the lowest energy excited state corresponds to an Ir - bpy charge-transfer excitation whereas in the crystalline host lattice [Rh(ppy)2bpy]PF6 the lowest excited state at 21 450 cm-1 is assigned to a 37r-r* excitation localized on the cyclometalating ppy- ligands. The next higher excited Ir - bpy charge-transfer state has shifted to 21 820 cm-', only 300 cm-I above the 3LC excited state. The close proximity of the 3LC and 3MLCT excited states and the large spin-orbit coupling constant of Ir3+ induce a strong mixing of charge-transfer character into the 3LC lowest excited states, resulting in increased oscillator strengths, reduced lifetimes, short axis polarized transitions, and a large zero-field splitting of 10-15 cm-1. |
|
| |  | |
-
Cooperative Formation of Inorganic-Organic Interfaces in the Synthesis of Silicate Mesostructures
A. Monnier, F. Schüth, Q. Huo, D. Kumar, D. Margolese, R.S. Maxwell, G.D. Stucky, M. Krishnamurty, P. Petroff, A. Firouzi, M. Janicke and B.F. Chmelka
Science, 261 (5126) (1993), p1299-1303
DOI:10.1126/science.261.5126.1299 | Abstract | Article PDF
|
A model is presented to explain the formation and morphologies of surfactant-silicate mesostructures. Three processes are identified: multidentate binding of silicate oligomers to the cationic surfactant, preferential silicate polymerization in the interface region, and charge density matching between the surfactant and the silicate. The model explains present experimental data, including the transformation between lamellar and hexagonal mesophases, and provides a guide for predicting conditions that favor the formation of lamellar, hexagonal, or cubic mesostructures. Model Q230 proposed by Mariani and his co-workers satisfactorily fits the x-ray data collected on the cubic mesostructure material. This model suggests that the silicate polymer forms a unique infinite silicate sheet sitting on the gyroid minimal surface and separating the surfactant molecules into two disconnected volumes. |
|
|
Making use of the phenomenon of light-induced spin-crossover in the [Zn1âxFex(ptz)6](BF4)2 spin-crossover system, very high diffraction efficiencies η can be achieved in non-degenerate four-wave-mixing. In the mixed crystal with x=0.1 and at 76 K, i.e. at a temperature where the system is predominantly in the low-spin state, a value for η of 43% was obtained. This is attributed to a phase grating due to the large difference in metal---ligand bondlength between the low-spin ground state and the light-induced high-spin state. The rate constant for the decay of the laser-induced grating as a function of temperature is found to be exactly twice the one of the high-spinâlow-spin relaxation, as expected for a dilute system in the absence of cooperative effects. |
|
|
The two title compounds were synthesized and investigated with the inelastic-neutron-scattering (INS) technique. They contain mixed YbMBr93- (M=Cr3+, Ho3+) dimers as discrete units, and the magnetic excitations of mixed Yb3+-Cr3+ and Yb3+-Ho3+ dimers could thus be observed. The Yb3+-Cr3+ dimer has three INS transitions, for which anisotropic exchange, as well as zero-field splitting of Cr3+, has to be included in the exchange Hamiltonian. For the Yb3+-Ho3+ dimer the effect of the exchange interaction manifests itself as a broadening and a splitting of the crystal-electric-field levels of the isolated Ho3+ ion. Taking into account the full (2JÂ + 1) ground-state multiplet of Ho3+, as well as anisotropic exchange, gives a satisfactory description of this dimer. |
|
|
We present the results of a crystallographic and optical study realized on the system SryBa1-yFClxBr1-x where x and y vary from zero to one. All mixed crystals studied were of tetragonal symmetry with the parent PbFCl structure. Complete structure determinations were performed for two single crystals with y=0.3 and x=1 respectively 0.7 (nominal composition). This system forms complete solid solutions, in contrast to previously published results. |
|
|
Spectral hole-burning studies of nile red and cresyl violet in polyvinylbutyral and polyvinylformal films have been performed. From the shape of spectral holes under the influence of an electric field, the dipole moment difference between the ground and excited state of both dyes has been determined. The Stark effect was investigated at different positions in the inhomogeneously broadened absorption band of the guest molecules. The observed dipole moment difference decreases with increasing wavelength. This variation is caused by the matrix induced dipole moment. For nile red, which is a neutral and polar molecule, the distribution of induced dipole moments is strongly correlated with the orientation of its ground state dipole moment. In the case of cresyl violet perchlorate, which is a salt, this distribution is anisotropic for guests absorbing in the blue part of the inhomogeneous band but becomes more isotropic as the absorption wavelength increases. The wavelength dependence of the observed dipole moment is much stronger and is ascribed to the existence of the cresyl violet perchlorate salt in different states of solvation. |
|
|
Holographic detection of spectral holes is demonstrated in a crystalline host material with signal-to-noise ratios of up to 104. Hole burning occurs in two Pr3+ sites in the Y2SiO5 lattice, in both cases due to population redistribution between the ground-state quadrupole levels. The signal contains contributions due to a resonant hole and several side holes and antiholes, a phenomenon not previously observed using the holographic technique. The diffracted spectrum is modeled in two ways. In the first case the transmission spectrum is used to determine the population gratings and thus the diffraction efficiency. In the second case the transition probabilities between ground- and excited-state Kramer's doublets are used to model the population gratings. The technique is applied to pseudo-Stark-effect measurements from which the crystallographic sites as determined by x-ray analysis are matched to the spectroscopic data presented here. The time decay of the diffracted signal is used to study nuclear spin-lattice relaxation. It is shown that at 1.6 K temperature-dependent phonon-induced processes make no contribution to this decay. The nonexponential time decay of the population upon radio-frequency irradiation resonant with a ground-state quadrupole splitting is attributed to Pr-Pr cross relaxation |
|
|
A detailed study of the separation efficiency in the photoinduced electron transfer reaction between 9,10-dicyanoanthracene and biphenyl in acetonitrile is presented. Both transient absorption and photoconductivity indicate a separation efficiency of about 0.4. This value is in discrepancy with two of three previously reported efficiencies. The problems arising with too large donor concentrations and with the use of a secondary donor to determine the separation efficiency are discussed. |
|
|
A study of the hole-burning mechanisms of bis[4-(diethylamino)-2-hydroxyphenyl]squaraine (DEAH) and bis[4-(disethylamino)-phenyl]squaraine (DEA) in hydrogen-bonding and non-hydrogen-bonding polymers is presented. Intramolecular H-bonding is only possible for DEAH. In all systems, the spectral holes are not persistent and decay with a distribution of rates ranging from 10-5s-1 to about 1 s-1, the time resolution of the experiment. In H-donating matrices, this distribution varies with the burning wavelength. From the hole-burning efficiencies and the kineticsof the hole refilling, four different types of nonphotochemical hole-burning mechanisms are postulated. The efficiency of these mechanisms depends mainly on the occurrence of processes slowing down the relaxation to the initial product state. |
|
|
The rotational dynamics of nile red has been studied in polar protic, polar aprotic and non-polar solvents. In the non-polar and the aprotic solvents, with the exception of long alkanenitriles, the rotation dynamics is consistent with the prediction of the StokesâEinsteinâDebye hydrodynamics theory for slip and close to the stick boundary condition, respectively. However in protic solvents, the rotation dynamics can be explained in terms of the StokesâEinsteinâDebye hydrodynamics theory under stick boundary condition only if solvent attachment via hydrogen bonding is assumed. The anomalous behaviour observed in longer alkanenitriles has been assigned to the formation of a reverse micelle-like solvation layer around nile red. |
|
|
The conformational analysis of TMABN by three different methods X-ray analysis, photoelectron spectroscopy, and UV molar absorption coefficient yields a twist angle of the dimethylamino group of 60-70° in the ground state, whereas DMABN is not far from planar in qualitative agreement with the predictions from force field calculations (QCFF/PI and MM3). Dipole moment determinations by the thermochromic method agree with those from other methods (solvatochromism, electrochromism and time resolved microwave absorption) in that the excited state dipole moment of TMABN is very large, as well as that of the TICT state of DMABN. Its value increases somewhat with solvent polarity. This is explained by a nuclear polarizability model. The force field calculations are used to predict twist angle values for various sterically hindered DMABN derivatives |
|
|
Quantum chemical calculations based on density functional theory have been performed on ruthenocene. Excellent agreement is obtained with groundâ and excitedâstate properties derived from optical spectroscopy. In particular, the energies of the first dâd excitations, the unusually large Stokes shift, the structural expansion of Ru(cp)2 and the substantial reduction of the Ruâcp force constant in the first triplet excited state are almost quantitatively reproduced. The lowestâenergy excitation is found to have substantial charge transfer character. |
|
|
Copper and silver, respectively, were introduced into single crystals of CsCdF3. Our detailed electron paramagnetic resonance (EPR) study showed that both elements enter the Cd lattice siteâcopper as Cu2+, silver as Ag+, which then was converted into Ag2+ by x raying the corresponding samples. Cu2+ and Ag2+ were shown to present in their ground state a pseudostatic JahnâTeller effect. Motional effects were observed in the respective EPR spectra and studied in some detail for Cu2+ as they are seen over a wide temperature range. Predictions of a stochastic Kubo model [J. Phys. Soc. Jpn. 9, 935 (1954)] were compared with the temperature dependent linewidths of the motionally averaged EPR spectrum. A power law (Tn with nâ1.9) was determined for the temperature dependence of the reorientation frequency between 30 and 90 K. |
|
|
Ab initio electronic structure calculations for phenol and the hydrogen-bonded complexes phenol · H2O and d-phenol · D2O were performed at the Hartree-Fock 4-31G and 6-31G** levels. Both phenol and phenol · H2O were fully structure optimized. Based on the minimumenergy structures so obtained, full normal coordinate analyses were carried out. The resulting harmonic frequencies were scaled and compared to available experimental data. The agreement is satisfactory and allows for an assignment of a majority of the bands observed in the experimental spectra. Comparison with previous calculations on (H2O)2 reveals a considerable increase in the strength of the hydrogen bond on going from (H2O)2 to phenol · H2O. |
|
-
Novel triterpene-derived hydrocarbons of the arborane/fernane series in sediments: Part II
V. Hauke, R. Graff, P. Wehrung, J.M. Trendel, P. Albrecht, A. Riva, G. Hopfgartner, F.O. Gülaçar, A. Buchs and P.A. Eakin
Geochimica et Cosmochimica Acta, 56 (9) (1992), p3595-3602
DOI:10.1016/0016-7037(92)90405-8 | Abstract | Article PDF
|
|
|
A practical ab initio quantum-mechanical approach for calculations of free energies of molecules in solutions is developed. This approach treats the solute molecules by an explicit ab initio self-consistent-field approach while representing the solvent molecules by a pseudopotential. The solvation energies are evaluated by a free-energy perturbation approach that uses the distribution function associated with a classical force field as a reference state for the quantum-mechanical calculations. The performance of the method is examined by evaluating the solvation energy of an Li + ion. It is found that the calculation times are not much longer than that of the corresponding classical free-energy perturbation calculations. |
|
|
Ab initio selfâconsistentâfield (SCF) HartreeâFock and configuration interaction (CI) calculations have been carried out for H+ 2n+1 (n=1â6) clusters using a tripleâzeta plus polarization basis set. Fully optimized structures and energies of H+ 11 and H+ 13 are presented. These structures can be thought as the addition of H2 molecules to a deformed H+9. Dissociation energies as a function of cluster size follow the pattern established experimentally by Hiraoka and Mori. Nevertheless, our energy results on the biggest clusters suffer from the lack of size consistency of CI with single and double substitutions (CISD) calculations. Analytic gradient techniques have been used to locate stationary point geometries and to predict harmonic vibrational frequencies and infrared intensities at the two levels of theory SCF (n=1â6) and CISD (n=1â4) both with tripleâzeta polarizationbasis sets. Of special interest are the new vibrational modes of H+ 11 and H+ 13, which have no counterpart in the H+9 cluster. Our predicted frequencies compare fairly well with the experimental results of Okumura, Yeh, and Lee. |
|
-
Members of the PbFCl-type family : possible candidates for room-temperature photochemical hole burning
R. Jaaniso, H. Hagemann, F. Kubel and H. Bill
Chimia, 46 (4) (1992), p133-137
unige:2908 | Abstract | Article PDF
|
We report on crystal growth and about physico-chemical studies on SryBa1âyFClxBr1âx (y = 0, 0.5, and 1) compounds doped with Sm. Persistent spectral hole burning at 300 K is further reported on Sr0.5Ba0.5FCl0.5Br0.5:Sm single crystals. |
|
|
The high-spin to low-spin (HSâLS) relaxation in the [Fe(ptz)6](BF4)2 spin-crossover system deviates strongly from first-order kinetics because of cooperative effects of elastic origin. The shift in horizontal and vertical displacement of the potential wells of the initial and final state relative to each other due to the build-up of an "internal" pressure is estimated from spectroscopic measurements. The HSâLS relaxation as such is described by the theory of nonadiabatic multiphonon relaxation in the strong-coupling limit, with a HuangâRhys factor S â 45 which is much larger than the reduced energy gap p. The sigmoidal relaxation curves in [Fe(ptz)6](BF4)2 result when a change in S of â â1 and in p of 1 during the relaxation is taken into account. |
|
|
The constructive interference between two Stark-effect-broadened holograms produced by spectral hole burning is discussed. The holograms are burned at the same frequency but at different external electric-field values. The phase difference is selected to be zero so that constructive interference between the waves diffracted by each grating occurs. Experimentally it is found that a dip in the hologram efficiency that is not predicted by previous theory occurs for all reconstruction external electric-field values in the region between the original burn values. This dip is interpreted as being due to the time nonlinearity of the hologram burn process. The dip corresponds to those molecules, oriented in a specific direction with respect to the electric field, for which no Stark shift occurs and that are therefore resonant with the laser during the production of both holograms. The width of the anomalous feature is close to that of the hologram when the hologram is reconstructed at the original burn external electric-field strength. Other molecular orientations may be selected by burning pairs of holographic gratings at other combinations of the frequency and the electric field. |
|
|
The electron transfer reaction between 9,lO-anthraquinone (AQ) excited to the lower triplet state and 1,2,4trimethoxybenzene (TMB) in solvents of different polarity has been studied by nanosecond time-resolved resonance Raman spectroscopy. In no solvent was there evidence for the formation of a triplet exciplex. All the observed vibrations were characterized as AQ* or TMB" modes. The frequency of the band assigned to the C-C stretch of TMB" has been found to be very dependent upon the environment and this effect has been used to observe the separation of the geminate ion pair into free ions in polar solvents. The data strongly suggest that the ions are at van der Waals contact in the geminate ion pair and not separated by solvent molecules. |
|
-
Experimental and theoretical investigation of asymmetric induction in the synthesis of disubstituted cyclohexadienes via chiral benzene chromium complexes
G. Bernardinelli, A.F. Cunningham, C. Dupré, E.P. Kündig, D. Stussi and J. Weber
Chimia, 46 (4) (1992), p126-129
unige:2932 | Abstract
|
A series of [Cr(benzene)(CO)2L] complexes with L = PPh3, P(OMe)3, PPh2 ((â)-menthyl), P(OPh)2(O-(â)-menthyl), P(O-(â)-menthyl)3 were subjected to a nueleophile addition/acylation sequence to give trans-5,6-disubstituted cyelohexadienes. Low-to-moderate asymmetric induction was observed with the chira] ligands. Experimental and theoretical evidence for an alkylation at the metal center trans to the P ligand is presented, and a crystal structure determination of a [Cr(η5-cyclohexadienyl)(P(OMe)3)(CO)2SnPh3] complex is included. |
|
|
The ion Ag2+ introduced into NaF shows a tetragonal electron paramagnetic resonance spectrum at 4.2 K which dynamically averages above â40 K. Uniaxial stress is used to show that the ground state is a strongly coupled Eâϵ JahnâTeller state. The wellâresolved superhyperfine structure due to the Fâ neighbors is analyzed with a linear combination of atomic orbitals picture. Optical absorption of asâgrown and treated crystals is further presented. The former ones show peaks at 202, 213, 219 nm due to Ag+. The latter ones present complex absorption spectra related to silver. |
|
|
The 5T2(HS)1A1(LS) intersystem crossing rates have been determined for a number of Fe(II) coordination compounds between 10 and 270 K using time-dependent optical spectroscopy. Strong deviations from Arrhenius kinetics with nearly temperature independent tunneling at low temperatures and a thermally activated behavior at elevated temperatures with apparent activation energies smaller than the classical energy barrier were found. The tunneling rates range from ~10â6 sâ1 for the doped spin crossover system [Zn1âxFex(ptz)6](BF4)2 to ~106 sâ1 for the doped low-spin (LS) system [Zn1âxFex(bipy)3](PF6)2. The large range of 12 orders of magnitude in the low temperature tunneling rates as well as the activated region can be understood in terms of nonadiabatic multiphonon relaxation. Values for the HuangâRhys parameter S of 40â50 and for the reduced energy gap p of 1â12 are estimated for the present series of compounds. The validity of an inverse energy gap law in the strong vibronic coupling limit with Sp is borne out by experiment. |
|
|
The temperature dependent Raman spectra of the title compound confirm the presence of some rotational disorder of the NH3Â end groups below 112 K. The central carbon-carbon stretching mode around 865 cm-1Â is coupled to the order parameter of the incommensurate phase transition at 168 K. No other clear evidence of the incommensurate modulation appears in the Raman spectra between 112 and 168 K. In the Abma phase (above 168 K) a dynamic conformational gauche-trans equilibrium is observed. The corresponding enthalpy difference is estimated to be 18.4+or-6.5 kJ mol-1. |
|
|
Due to the fact that for d6systems there are a number of low-lying ligand field (LF) states the relaxation from excited states of Fe(II) coordination compounds is, in general, a very fast and radiationless process. In Fe(II) spin-crossover systems, however, the zero point energy difference between the two lowest states, namely the low-spin (LS) 1A1 and the high-spin (HS) 5T2 state, is of the order of kBT, and some systems can be converted quantitatively to the HS state well below the thermal transition temperature by irradiating either into MLCT or LF absorption bands of the LS species, with HSâLS relaxation rates as small as 10â6 sâ1 at XXX10 K. It is also possible to achieve a light-induced transient population of a HS state in Fe(II) LS compounds, but in this case the HSâLS relaxation rates can be larger than 106 sâ1 even at low temperatures. The HSâLS relaxation rates show strong deviations from Arrhenius kinetics with nearly temperature independent tunnelling below Ë70 K and a thermally activated behaviour above Ë100 K. The range of 12 orders of magnitude in the low temperature tunnelling rate can be understood in terms of nonadiabatic multiphonon relaxation, where in the strong coupling limit, with the Huang-Rhys parameter S much larger than the reduced energy gap p, an inverse energy gap law holds. |
|
|
[Fe(ptz)6](BF4)2 (ptz=1-propyltetrazole) is an Fe(II) spin crossover system, which shows a light-induced low-spin (1A1)-->high-spin (5T2) conversion below ~50 K by irradiating into the spin allowed 1A1-->1T1 dâd absorption band. This phenomenon, known as light-induced excited spin state trapping (LIESST), is reversible, and a subsequent irradiation into the 5T2-->5E band results in a light-induced 5T2-->1A1 conversion (reverse LIESST). Single crystal absorption spectra of the title compound in the region of dâd transitions are reported. In addition to the well-established spin allowed 1A1-->1T1 and 1A1-->1T2 transitions of the low-spin species and the 5T2-->5E transition of the high-spin species two weak bands in the NIR are assigned to the spin forbidden 1A1-->3T1 and 1A1-->3T2 transitions. Direct irradiation into the 1A1-->3T1 absorption band at 20 K results in a quantitative 1A1-->5T2 conversion, proving that this low lying triplet state plays an important role in the mechanism of LIESST. A full kinetic scheme for LIESST and reverse-LIESST with the 3T1 state as intermediate state is developed, and the quantum efficiencies for the various intersystem crossing steps involved are given: they are of the order of unity for the first step from the initially excited 1T1 and 5E states to the intermediate 3T1 state, respectively. The branching ratio from the 3T1 state to the 1A1 and the 5T2 states is 1:4. |
|
|
The observatiqn of photon-gated hologram formation in a boric acid glass doped with triphenylene is reported. The first photon excites triphenylene to its first singlet excited state and, through intersystem-crossing, populates the first triplet stateTI. The second photon excites TI to T,, where autoionization occurs, leading to the formation of a radical cation. The gatinglight populating TI via SI is spatially uniform, while the light exciting TI to T, is spatially modulated. The long lifetime of the first triplet state allows Recording with low light intensities (mW/cm2). The spatially modulated excitation light forms three gratings (educt, intermediate state, and product). The extent of the interaction between these gratings depends on the overlap between educt, intermediate, and product absorption and refraction spectra as well as on the reading wavelength. The holograms were read at 363.8 and 632.8 nm. When the gating light is blocked, the holographic efficiency stays constant when read at 632.8 nm but increases substantially when read at 363.8 nm. |
|
|
In conditions of laser flash photolysis, the kinetics of decay of the absorption of the benzophenone radical anion show that free, solvated ions are formed after electron transfer between the title compounds in neat, dry acetonitrile. Furthermore, it is shown that the opposite conclusion claimed by Devadoss and Fessenden (J. Phys. Chem., 1990, 94,4540), Le., no ion pair dissociation, results from a misinterpretation of the transient decay rate. |
|
|
It is shown that using an ESR spectrometer with magnetic field modulation and sweeping the temperature across Tc (at a constant and a very low magnetic field), is equivalent to temperature modulation. The signal intensity obtained when crossing Tc is proportional to 1/( delta Hc2/ delta T) at T=Tc. Using the WHH relation Hc2(T=0)=0.7 Tc( delta Hc2/ delta T)T=Tc enabled the measurement of the relative angular variation of Hc2 in single crystals of YBaCuO with Tc approximately 85 K. The data fit the Ginzberg-Landau theory. This very sensitive technique can be used to characterize properties of high Tc superconducting materials. Results on thin films is also be presented. |
|
|
|  |
It is shown that measuring microwave absorption in high-Tc superconductors at constant and very low magnetic fields, using magnetic-field modulation, is, under some conditions, equivalent to temperature modulation when sweeping the temperature across Tc. Using an ESR spectrometer, the derivative of microwave absorption is measured close to Tc. This allows a determination of the relative angular variation of dHc2/dT at T=Tc in single crystals of Y-Ba-Cu-O. The data fit the Ginzburg-Landau theory on the relative angular variation of Hc2. The ratio (dHc2/dT)T=Tc parallel and perpendicular to the Cu planes was found to be 2.7 and 5.3 for two Y-Ba-Cu-O single crystals with Tc=89 and 86 K, respectively. These values obtained at 1010 Hz are close to the values obtained by conventional dc methods. |
|
|
Recently, we have discovered a fascinating photophysical effect in spin crossover complexes of iron(II) : Light-Induced Excited Spin State Trapping (LIESST). At sufficiently low temperatures, the low spin state (1A1) can be converted quantitatively to the high spin state (5T2) by irradiating the sample into the 1A1 â 1T1 d-d absorption band (â540 nm). The resulting metastable HS state has a very long lifetime at low temperatures, in some cases it does not decay noticeably over a period of several days at 10 K. Only at temperature above some critical temperature does thermal relaxation back to the LS state set in. The sample can also be reconverted to the LS state by irradiating into the 5T2 â 5E absorption band (â50 nm). The system thus behaves like an optical switch. The relative positioning - horizontally and vertically - of the potential wells of the two spin states is crucial for the lifetime of the metastable HS state. |
|
|
Polarized Raman measurements on single crystals of CuO yield the symmetry assignments of the three predicted Raman active lattice modes : 297cmâ1 (Ag, 344cmâ1 (Bg) and 629cmâ1 (Bg). These results are compared to literature data, including IR spectra. Our measurements confirm at low temperature the appearance of an additional Raman band around 240 cmâ1. The temperature dependence of the linewidth of the Ag mode presents an anomalous behavior near the magnetic phase transition, suggesting the possible presence of magnon-phonon couplings in the antiferromagnetic phase. |
|
|
We report on the growth of Nd2-xCexCuO4-δ single crystals (0<x<0.2) from Cu2O flux. Free separated crystals with maximum size of 5x8x0.15 nm3 have been obtained. Magnetic AC susceptibility measurements show a sharp superconducting transition at temperatures up to 23 K. The temperature dependence of the lattice parameters has been measured by means of X-ray powder diffraction between 10 K (a=3.9413(3) Ã
, c=12.0290(18)Â Ã
) and 290 K (a=3.9482(3)Â Ã
, c=12.0590(18)Â Ã
). Room temperature Raman spectra reveal a new band at 320 cm-1 which is not observed in Nd2CuO4. Raman spectra of crystals withTc ranging from 7 to 22 K show a systematic intensity change of the broad band at 590 cm-1. |
|
|
The polarized Raman spectra of four different beryl crystals were studied at room temperature in the range from 30 to 4000 cm-1. The spectra show significant differences between the samples studied, and corrections are proposed for the reference Raman spectra of beryl previously reported by Adams and Gardner (1974). Type II water is observed in two crystals; the corresponding symmetric Raman stretching band at 3595 cm-1Â is extremely strong for an impurity (about 20% of the strongest beryl lattice mode). Another, sharper, band of similar intensity at 3605 cm-1Â could possibly originate from a hydroxyl stretching mode. Additional weaker bands are observed around 1600 cm-1Â and 3600â3750 cm-1. The first polarized Raman spectra of bazzite are presented and discussed. |
|
|
A study of viscosity and temperature effects on the rate of back electron transfer (BET) within an exciplex (9,10-dicyanoanthracene/N,N-dimethylaniline) with a strong charge transfer character in six non-polar solvents is reported. The extent of charge transfer has been estimated from the solvatochromic and thermochromic shifts of the fluorescence. Conformational changes are a prerequisite to the BET. In non-viscous solvents, where they are much faster than the ET step itself, the observed rate can be explained within the theory of non-adiabatic ET reactions, while in more viscous solvents, a time-dependent electronic coupling constant V has been introduced. In decalin and butylbenzene, a transition from a "solvent independent" to a "solvent controlled" non-adiabatic regime is observed. |
|
| | 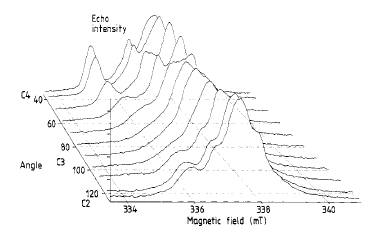 | |
|
The construction of a new high-resolution multichannel low-cost pulse generator is reported. It is fully computer controlled through a simple RS232 serial interface. Its features are 1 ns resolution within any time, pulse delays up to 16 ms and pulse lengths up to 65 mu s, and pulse sequence repetition rate from 66 Hz up to 250 kHz. It has fully programmable sequencing, including step increments for any pulse delay or length. It governs a pulsed ESR spectrometer, which is also described, but it could be used in a very wide range of experimental set-ups. A few examples of spin-echo detected ESR and ESEEM of some paramagnetic centres are shown. |
|

