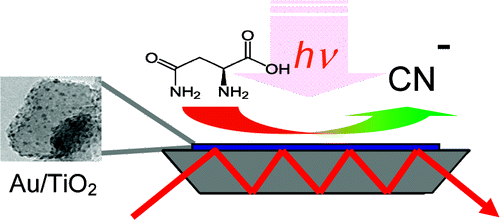
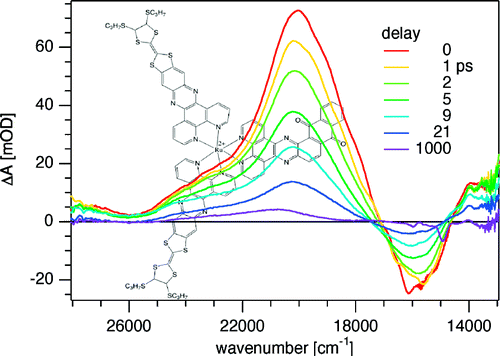
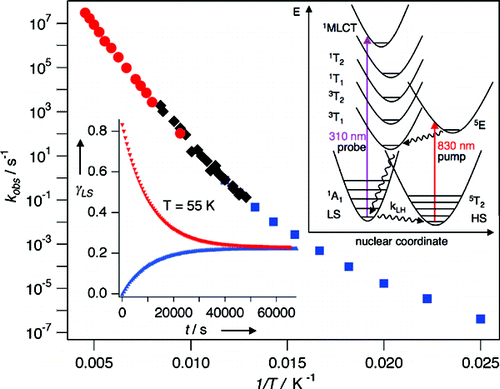
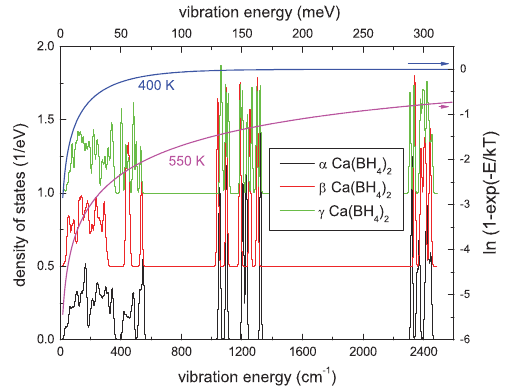
Over recent years, research on thiolate-protected gold clusters Aum(SR)n has gained significant interest. Milestones were the successful determination of a series of crystal structures (Au102(SR)44, Au25(SR)18, Au38(SR)24, Au36(SR)24, and Au28(SR)20). For Au102(SR)44, Au38(SR)24, and Au28(SR)20, intrinsic chirality was found. Strong Cotton effects (circular dichroism, CD) of gold clusters protected by chiral ligands have been reported a long time ago, indicating the transfer of chiral information from the ligand into the cluster core. Our lab has done extensive studies on chiral thiolate-protected gold clusters, including those protected with chiral ligands. We demonstrated that vibrational circular dichroism can serve as a useful tool for the determination of conformation of the ligand on the surface of the cluster. The first reports on crystal structures of Au102(SR)44 and Au38(SR)24 revealed the intrinsic chirality of these clusters. Their chirality mainly arises from the arrangement of the ligands on the surface of the cluster cores. As achiral ligands are used to stabilize the clusters, racemic mixtures are obtained. However, the separation of the enantiomers by HPLC was demonstrated which enabled the measurement of their CD spectra. Thermally induced inversion allows determination of the activation parameters for their racemization. The inversion demonstrates that the goldâthiolate interface is anything but fixed; in contrast, it is rather flexible. This result is of fundamental interest and needs to be considered in future applications. A second line of our research is the selective introduction of chiral, bidentate ligands into the ligand layer of intrinsically chiral gold clusters. The ligand exchange reaction is highly diastereoselective. The bidentate ligand connects two of the protecting units on the cluster surface and thus effectively stabilizes the cluster against thermally induced inversion. A minor (but significant) influence of chiral ligands to the CD spectra of the clusters is observed. The studied system represents the first example of an intrinsically chiral gold cluster with a defined number of exchanged ligands, full control over their regio- and stereochemistry. The methodology allows for the selective preparation of mixed-ligand cluster compounds and a thorough investigation of the influence of single ligands on the clusterâs properties. Overall, the method enables even more detailed tailoring of properties. Still, central questions remain unanswered: (1) Is intrinsic chirality a ubiquitous feature of thiolate-protected gold clusters? (2) How does chirality transfer work? (3) What are the applications for chiral thiolate-protected gold clusters? In this Account, we summarize the main findings on chirality in thiolate-protected gold cluster of the past half decade. Emphasis is put on intrinsically chiral clusters and their structures, optical activity, and reactivity.
The Au102(p-MBA)44 cluster (p-MBA: para-mercaptobenzoic acid) is observed as a chiral compound comprised of achiral components in its single-crystal structure. So far the enantiomers observed in the crystal structure are not isolated, nor is the circular dichroism spectrum known. A chiral phase transfer method is presented which allows partial resolution of the enantiomers by the use of a chiral ammonium bromide, (â)-1R,2S-N-dodecyl-N-methylephedrinium bromide ((â)-DMEBr). At sufficiently low concentration of (â)-DMEBr, the phase transfer from water to chloroform is incomplete. Both the aqueous and organic phases show optical activity of near mirror image relationship. Differences in the spectra are ascribed to the formation of diastereomeric salts. At high concentrations of (â)-DMEBr, full phase transfer is observed. The organic phase, however, still displays optical activity. We assume that one of the diastereomers has very strong optical activity, which overrules the cancelation of the spectra with opposite sign. Comparison with computations further corroborates the experimental data and allows a provisional assignment of handedness of each fraction.
Surface-enhanced Raman spectroscopy takes advantage of plasmonic substrates that sustain resonances at tunable frequencies with a reproducibly extraordinary field enhancement. Low-cost and large-scale fabrication of these substrates is further required. Here, we present stacked large-scale arrays of strongly coupled gold nanoparticles as promising candidates for such substrates. These arrays are fabricated by bottom-up techniques that fulfill the aforementioned requirements. The distance between adjacent arrays in the stack is controlled with high precision using a discrete number of monolayers of molecules that enable the spectral position of the plasmonic resonances to be tuned. Although the nanoparticles are randomly arranged in each array, the spatial proximity of the stacked arrays enables a strong coupling among nanoparticles to be achieved in adjacent arrays. The huge field enhancements due to these strongly coupled gold nanoparticles are shown to enhance the Raman signal. We show that effectively the optical response from these stacked arrays and the Raman signals can be understood in a simplifying picture where only an individual nanoparticle dimer is considered. The possibility to tune the plasmonic resonances of the substrate across the visible spectrum makes our material a plasmonic substrate of choice for many applications where lightâmatter interactions need to be intensified.
A simple method is presented to control and trigger the coupling between plasmonic particles using both a growing process of gold nanoparticles (GNPs) and a mechanical strain applied to the elastomeric template where these GNPs are anchored. The large scale samples are prepared by first depositing and then further growing gold nanoparticles on a flexible PDMS tape. Upon stretching the tape the particles move further apart in the direction of the stretching and closer together in the direction perpendicular to it. The synergy between the controlled growth of GNPs and the mechanical strain, leads to a drastic shift of the plasmon band and a color change of the sample. Furthermore, the stretching by only a few percent of the amorphous and initially isotropic sample results in a strong polarization-dependent plasmon shift. At smaller gap sizes between neighboring particles, induced by stretching the PDMS tape, the plasmon shift strongly deviates from the behaviour expected considering the plasmon ruler equation. This shows that multipolar coupling effects significantly contribute to the observed shift. Overall, these results indicate that a macroscopic mechanical strain allows one to control the coupling and therefore the electromagnetic field at the nanoscale.
The most abundant of the modified nucleosides, and once considered as the âfifthâ nucleotide in RNA, is pseudouridine, which results from the action of pseudouridine synthases. Recently, the mammalian pseudouridine synthase 1 (hPus1p) has been reported to modulate class I and class II nuclear receptor responses through its ability to modify the Steroid receptor RNA Activator (SRA). These findings highlight a new level of regulation in nuclear receptor (NR)-mediated transcriptional responses. We have characterised the RNA association and activity of the human Pus1p enzyme with its unusual SRA substrate. We validate that the minimal RNA fragment within SRA, named H7, is necessary for both the association and modification by hPus1p. Furthermore, we have determined the crystal structure of the catalytic domain of hPus1p at 2.0 Ã resolution, alone and in a complex with several molecules present during crystallisation. This model shows an extended C-terminal helix specifically found in the eukaryotic protein, which may prevent the enzyme from forming a homodimer, both in the crystal lattice and in solution. Our biochemical and structural data help to understand the hPus1p active site architecture, and detail its particular requirements with regard to one of its nuclear substrates, the non-coding RNA SRA.
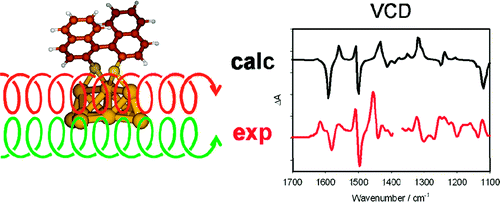
Cationic azaoxa[4]helicenes can be prepared in a single step from a common xanthenium precursor by addition of nucleophilic amines under monitored conditions (160 °C, 2 min, MW). The (â)-(M) and (+)-(P) enantiomers can be separated by chiral stationary-phase chromatography. Determination of the absolute configuration and racemization barrier (ÎG⧧ (433 K) 33.3 ± 1.3 kcal·molâ1) was achieved by VCD and ECD spectroscopy, respectively.
Non-uniform Continuum Model for Solvated Species Based on Frozen-Density Embedding Theory: The Study Case of Solvatochromism of Coumarin 153
S.V. Shedge, X. Zhou and T.A. Wesolowski Chimia, 68 (9) (2014), p609-614 Keywords: continuum models; density functional theory; multi-level simulations; orbital-free embedding theory; solvatochromism
DOI:10.2533/chimia.2014.609 | unige:75040 | Abstract
Recent application of the Frozen-Density Embedding Theory based continuum model of the solvent, which is used for calculating solvatochromic shifts in the UV/Vis range, are reviewed. In this model, the solvent is represented as a non-uniform continuum taking into account both the statistical nature of the solvent and specific soluteâsolvent interactions. It offers, therefore, a computationally attractive alternative to methods in which the solvent is described at atomistic level. The evaluation of the solvatochromic shift involves only two calculations of excitation energy instead of at least hundreds needed to account for inhomogeneous broadening. The present review provides a detailed graphical analysis of the key quantities of this model: the average charge density of the solvent (<ÏB>) and the corresponding Frozen-Density Embedding Theory derived embedding potential for coumarin 153.
Crystal-clear - The '2014 Most Superlative Crystal Growth Contest' for School Classes
D. Perret, H. Hagemann, R. Cerny, C. Renner, E. Giannini, L. Guénée, C. Besnard, D. Gérard and L. Windels Chimia, 68 (12) (2014), p893-895 Keywords: crystal growth contest; crystallography and society; education; incentive science; interrnational year of crystallography 2014; school science; potassium dihydrogen phosphate; outreach initiative
DOI:10.2533/chimia.2014.893 | unige:46708 | Article PDF
To celebrate the International Year of Crystallography among the general public, a consortium of chemists, physicists and crystallographers of the University of Geneva organised in Spring 2014 an incentive crystal growth contest for Geneva scholars aged 4 to 19. Starting from a kit containing a salt and user instructions, classes had to prepare a crystal that met specific criteria according to their category of age. The composition of the salt â potassium dihydrogen phosphate (KDP) â was only disclosed to the participants during the final Awards Ceremony. This contest positively exceeded our expectations with almost 100 participating classes (ca. 1800 participants) and 54 specimens received over all categories.
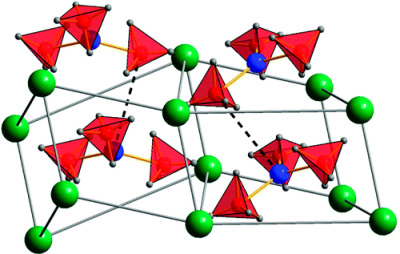
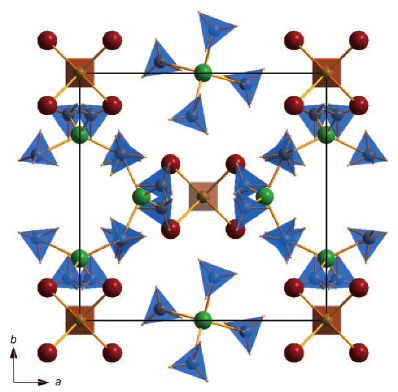
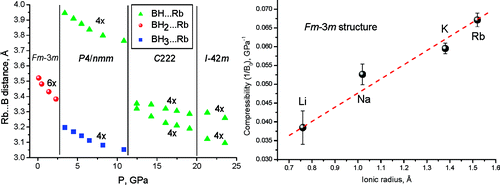
Perovskite materials host an incredible variety of functionalities. Although the lightest element, hydrogen, is rarely encountered in oxide perovskite lattices, it was recently observed as the hydride anion Hâ, substituting for the oxide anion in âBaTiO3. Here we present a series of 30 new complex hydride perovskite-type materials, based on the non-spherical âtetrahydroborate anion âBH4â and new synthesis protocols involving rare-earth elements. Photophysical, electronic and âhydrogen storage properties are discussed, along with counterintuitive trends in structural behaviour. The electronic structure is investigated theoretically with density functional theory solid-state calculations. BH4-specific anion dynamics are introduced to perovskites, mediating mechanisms that freeze lattice instabilities and generate supercells of up to 16 Ã the unit cell volume in AB(BH4)3. In this view, homopolar hydridic di-hydrogen contacts arise as a potential tool with which to tailor crystal symmetries, thus merging concepts of molecular chemistry with ceramic-like host lattices. Furthermore, anion mixing âBH4ââXâ (Xâ=Clâ, Brâ, Iâ) provides a link to the known ABX3 halides.
Excited State and Injection Dynamics of Triphenylamine Sensitizers Containing a Benzothiazole Electron-Accepting Group on TiO2 and Al2O3 Thin Films
M. Fakis, P. Hrobárik, O. Yushchenko, I. Sigmundová, M. Koch, A. Rosspeintner, E. Stathatos and E. Vauthey The Journal of Physical Chemistry C, 118 (49) (2014), p28509-28519
DOI:10.1021/jp509971q | unige:43537 | Abstract | Article HTML | Article PDF
The excited state and electron injection dynamics of three new organic sensitizers, comprising a triphenylamine moiety connected by an ethenylene (CâC double-bond) or ethynylene (CâC triple-bond) Ï-spacer to an electron-withdrawing benzothiazole bearing a cyanoacrylic acid anchoring group, have been studied using a combination of steady-state and femtosecond-resolved spectroscopies. The measurements were carried out for the three dyes in predominantly neutral and completely deprotonated forms in liquid solutions and bound on nanocrystalline TiO2 and Al2O3 thin films. In addition, quantum-chemical calculations were performed to predict absorption spectra of the sensitizers and their corresponding cation radicals. Time-resolved fluorescence (TRF) measurements on TiO2 indicate that electron injection takes place on a <0.2 ps time scale. Transient electronic absorption (TA) measurements provide evidence for the formation of radical cations not only in dye-sensitized TiO2 films but also in Al2O3 ones. The cation lifetime in Al2O3 is significantly shorter compared to TiO2, indicating a faster recombination of injected electrons with the dye cations. In addition, the ground-state bleach band in dye-sensitized TiO2 films experiences a gradual red-shift, which is indicative of a transient Stark effect. Finally, femtosecond transient absorption measurements in the IR region point to an ultrafast generation of injected electrons for all dyes. A faster recombination of the injected electrons with the dye cations is observed for the sensitizer decorated with auxiliary electron-donating methoxy groups on the triphenylamine moiety.
Single Crystal EPR Studies of Radicals Produced by Radiolysis of Organophosphorus Compounds
M. Geoffroy
in "Applications of EPR in Radiation Research"
Anders Lund - Masaru Shiotani Eds., Springer,
(2014), p33-66
DOI:10.1007/978-3-319-09216-4_2 | unige:42196
The main radical species produced by radiolysis of organophosphorus compounds are described in this chapter. Their identification is generally based on an analysis of the g and hyperfine tensors obtained from EPR experiments performed on irradiated single crystals. Special emphasis is placed on the properties of the 31P hyperfine tensor, which is often decisive in determining the structure of these radicals. Radiogenic species mentioned in the beginning of this review correspond to simple phosphorus-centered radicals (PR2, PR3â, PR4, PR3+, and R2PO). Then, more delocalized systems are reported (allylic structures, captodatively stabilized radicals, symmetrical radical ions containing a PâP bond). The effects of radiolysis on compounds containing low-coordinate phosphorus atoms (e.g. phosphaalkenes) are also described as well as the formation of radical pairs in irradiated phosphated sugars. The last part of the chapter deals with metallated radicals formed by radiolysis of metallic complexes M(CO)5P(H)Ph2 (with M = Mo, Cr, W). In some cases, phosphorus-centered radicals are compared with their arsenic analogues. For several systems the focus lies on dynamical effects; this is the case, for example, for the triptycenephosphinyl radical, which undergoes internal rotation around a PâC bond. Molecular rearrangements after radiolysis of some organophosphorus compounds (e.g. diphosphenes) are also reported.
Complementary Surface Second Harmonic Generation and Molecular Dynamics Investigation of the Orientation of Organic Dyes at a Liquid/Liquid Interface
D. Svechkarev, D. Kolodezny, S. Mosquera-Vázquez and E. Vauthey Langmuir, 30 (46) (2014), p13869-13876
DOI:10.1021/la503121g | unige:43539 | Abstract | Article HTML | Article PDF
The second-order nonlinear response of two dyes adsorbed at the dodecane/water interface was investigated by surface second harmonic generation (SSHG). These dyes consist of the same chromophoric unit, 2-pyridinyl-5-phenyloxazole, with an alkyl chain located at the two opposite ends. The analysis of the polarization dependence of the SSHG intensity as usually performed points to similar tilt angles of the two dyes with respect to the interface but does not give information on the absolute direction. Molecular dynamics (MD) simulations reveal that both dyes lie almost flat at the interface but have opposite orientations. A refined SSHG data analysis with the width of the orientational distribution yields tilt angles that are in very satisfactory agreement with the MD simulations.
Because of their key role in many areas of science and technology, bimolecular photoinduced electron transfer reactions have been intensively studied over the past five decades. Despite this, several important questions, such as the absence of the Marcus inverted region or the structure of the primary reaction product, have only recently been solved while others still remain unanswered. Ultrafast spectroscopy has proven to be extremely powerful to monitor the entire electron transfer process and to access, with the help of state-of-the-art theoretical models of diffusion-assisted reactions, crucial information like e.g. the intrinsic charge separation dynamics beyond the diffusion limit. Additionally, extension of these experimental techniques to other spectral regions than the UV-visible, such as the infrared, has given a totally new insight into the nature, the structure and the dynamics of the key reaction intermediates, like exciplexes and ions pairs. In this perspective, we highlight these recent progresses and discuss several aspects that still need to be addressed before a thorough understanding of these processes can be attained.
Copper-Catalyzed Propargylic Substitution of Dichloro Substrates: Enantioselective Synthesis of Trisubstituted Allenes and Formation of Propargylic Quaternary Stereogenic Centers
H. Li, D. Grassi, L. Guénée, T. Bürgi and A. Alexakis Chemistry - A European Journal, 20 (50) (2014), p16694-16706 Keywords: allenes;asymmetric catalysis;copper;Grignard reagents;P ligands
DOI:10.1002/chem.201404668 | unige:43540 | Abstract | Article HTML | Article PDF
An easy and versatile Cu-catalyzed propargylic substitution process is presented. Using easily prepared prochiral dichloro substrates, readily available Grignard reagents together with catalytic amount of copper salt and chiral ligand, we accessed a range of synthetically interesting trisubstituted chloroallenes. Substrate scope and nucleophile scope are broad, providing generally high enantioselectivity for the desired 1,3-substitution products. The enantioenriched chloroallenes could be further transformed into the corresponding trisubstituted allenes or terminal alkynes bearing all-carbon quaternary stereogenic centers, through the copper-catalyzed enantiospecific 1,1/1,3-substitutions. The two successive copper-catalyzed reactions could be eventually combined into a one-pot procedure and different desired allenes or alkynes were obtained respectively with high enantiomeric excesses.
An efficient and highly stereoselective fluorinative aza-semipinacol rearrangement is described. The catalytic reaction requires use of Selectfluor in combination with the chiral, enantiopure phosphate anion derived from acid L3. Under optimized conditions, cyclopropylamines A were transformed into β-fluoro cyclobutylimines B in good yields and high levels of diastereo- and enantiocontrol. Furthermore, the optically active cyclobutylimines were reduced diastereoselectively with L-Selectride in the corresponding fluorinated amines C, compounds of significant interest in the pharmacological industry.
Hydrolysis of metal borohydrides in the presence of CO2 has not been studied so far, although carbon dioxide contained in air is known to accelerate hydrogen generation. KBH4 hydrolysis promoted by CO2 gas put through an aqueous solution was studied by time-resolved ATR-FTIR spectroscopy, showing a transformation of BH4â into B4O5(OH)42â, and a drastically accelerated hydrogen production which can be completed within minutes. This process can be used to produce hydrogen on-board from exhaust gases (CO2 and H2O). We found a new intermediate, K9[B4O5(OH)4]3(CO3)(BH4)·7H2O, forming upon hydrolysis on air via a slow adsorption of the atmospheric CO2. The same intermediate can be crystallized from partly hydrolyzed solutions of KBH4 + CO2, but not from the fully reacted sample saturated with CO2. This phase was studied by single-crystal and powder X-ray diffraction, DSC, TGA, Raman, IR and elemental analysis, all data are fully consistent with the presence of the three different anions and of the crystallized water molecules. Its crystal structure is hexagonal, space group P-62c, with lattice parameters a = 11.2551(4), c = 17.1508(8) à . Formation of the intermediate produces 16 mol of H2 per mole of adsorbed CO2 and thus is very efficient both gravimetrically and volumetrically. It allows also for an elimination of carbon dioxide from exhaust gases.
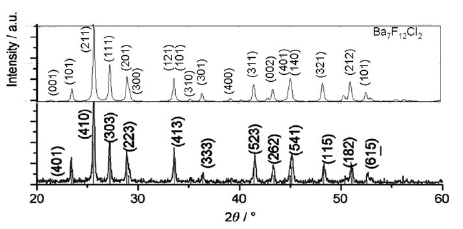
In analogy to the synthesis of polycrystalline M2NaIO6 (M = Ca, Sr, Ba) by precipitation in water at 90 °C, the title compound was first prepared as a metastable compound. The stable modification of Pb2NaIO6 was obtained by a heat treatment to 400 °C followed by cooling to room temperature. The crystal structure was refined from powder diffraction data [space group P21/c (14), a = 5.9040(2), b = 5.7526(2), c = 10.1104(3) à , β = 125.341(1)°]. On heating, at ca. 125 °C, a phase transition to a cubic high temperature modification was observed. The crystal structure was refined from XRD data measured at 200 °C [space group Fm3m (225), a = 8.2678(1) à ]. Depending on the precipitation temperature between 90 °C and 0 °C, several metastable modifications were obtained, which can be distinguished by significantly different lattice parameters. The XRD pattern of a powder precipitated at room temperature is pseudocubic. The crystal structure was refined at room temperature in P21/c with a = 5.8201(4), b = 5.8473(4), c = 10.0798(5) à , β = 125.074(3)°. This modification behaves almost as a cubic lattice on heating as found from XRD and DSC measurements.
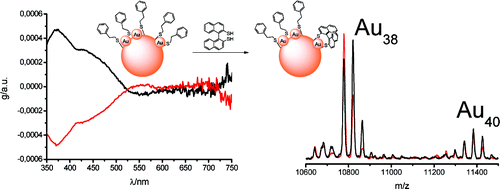
Pd2Au36(SC2H4Ph)24 clusters have been prepared, isolated and separated in their enantiomers. Compared to the parent Au38(SC2H4Ph)24 cluster the doping leads to a significant change of the circular dichrosim spectrum, however, the anisotropy factors are of similar magnitude in both cases. Isolation of the enantiomers allowed us to study the racemi-zation of the chiral cluster, which reflects the flexibility of the ligand shell composed of staple motifs. The doping leads to a substantial lowering of the racemization temperature. The change in activation parameters due to the doping may be solely due to modification of the electronic structure.
The compound Ba5I2O12 was synthesized by heating a precipitate of dissolved Ba(OH)2·8H2O and H5IO6. Rb2O was added to increase the crystallite size. The crystal structure was determined from conventional laboratory X-ray diffraction data by using a real-space structure solution approach followed by a Rietveld refinement. No constraints on positions were used. The structure analysis gave an orthorhombic symmetry with a = 19.7474(2) à , b = 5.9006(1) à and c = 10.5773(1) à . The final RBragg value in space group Pnma (62) was 1.0â%. The structure can be described by layers of a metal and iodine arrangement forming almost pentagonal holes. Raman measurements were correlated with the two IO6 octahedra. Two further barium periodate patterns were observed and indexed.
Where does the Raman optical activity of [Rh(en)3]3+ come from? Insight from a combined experimental and theoretical approach
M. Humbert-Droz, P. Oulevey, L.M. Lawson Daku, S. Luber, H. Hagemann and T. Bürgi Phys. Chem. Chem. Phys., 16 (42) (2014), p23260-23273
DOI:10.1039/C4CP02145B | unige:40863 | Abstract | Article HTML | Article PDF
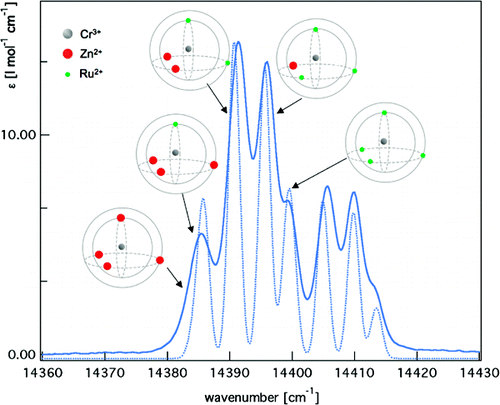
Backscattered Raman optical activity (ROA) spectra are measured for Î- and Î-tris-(ethylenediamine)rhodium(III) chloride in aqueous solution. In addition, the spectra of the four possible conformers in the Î configuration are investigated by ab initio calculations. The Î(δδδ) conformer is in best agreement with experimental spectra and examined in more details. The two most stable conformers according to the calculations are not compatible with the experimental ROA spectrum. Insights into the origin of observed band intensities are obtained by means of group coupling matrices. The influence of the first solvation shell is explored via an ab initio molecular dynamics simulation. Taking explicit solvent molecules into account further improves the agreement between calculation and experiment. Analysis of selected normal modes using group coupling matrices shows that solvent molecules lead to normal mode rotation and thus contribute to the ROA intensity, whereas the contribution of the Rh can be neglected.
Configurationally stable diaza[4]helicenes have been prepared in two steps by using a particularly facile NâN bond-cleavage reaction. The synthetic procedure uses hydrazine (NH2NH2) for the introduction of a single nitrogen atom. The strategy is general, modular and highly tolerant to functional groups. A mechanistic rationale is proposed for the spontaneous NâN bond-cleavage reaction. The resulting helical quinacridines are dyes that present absorption and emission properties that can be modulated as a function of pH; the pink quinacridine and green protonated forms (pKa â 9.0) display distinct optical features in the near-IR region. Single enantiomers were obtained by chiral stationary phase HPLC resolution. The absolute configurations were assigned by comparison of the ECD spectra of the conjugated acids with those of known dialkylquinacridinium derivatives. A rather high racemization barrier was measured by means of variable-temperature ECD experiments (ÎGâ¡ = 30.7â±â4.0 kcalâmolâ1 at 140 °C).
Nonuniform Continuum Model for Solvatochromism Based on Frozen-Density Embedding Theory
S.V. Shedge and T.A. Wesolowski ChemPhysChem, 15 (15) (2014), p3291-3300 Keywords: density functional calculations;molecular dynamics;molecular modeling;solvatochromism;UV/Vis spectroscopy
DOI:10.1002/cphc.201402351 | unige:41527 | Abstract | Article PDF
Frozen-density embedding theory (FDET) provides the formal framework for multilevel numerical simulations, such that a selected subsystem is described at the quantum mechanical level, whereas its environment is described by means of the electron density (frozen density; ÏB( r â) ) The frozen density ÏB( r â) is usually obtained from some lower-level quantum mechanical methods applied to the environment, but FDET is not limited to such choices for ÏB( r â). The present work concerns the application of FDET, in which ÏB( r â) is the statistically averaged electron density of the solvent <ÏB( r â)> . The specific soluteâsolvent interactions are represented in a statistical manner in <ÏB( r â)>. A full self-consistent treatment of solvated chromophore, thus involves a single geometry of the chromophore in a given state and the corresponding <ÏB( r â)>. We show that the coupling between the two descriptors might be made in an approximate manner that is applicable for both absorption and emission. The proposed protocol leads to accurate (error in the range of 0.05â eV) descriptions of the solvatochromic shifts in both absorption and emission.
We report herein on the polymer-crystallization-assisted thiol-ene photosynthesis of an amphiphilic comb/graft DNA copolymer, or molecular brush, composed of a hydrophobic poly(2-oxazoline) backbone and hydrophilic short single-stranded nucleic acid grafts. Coupling efficiencies are above 60% and thus higher as compared with the straight solid-phase-supported synthesis of amphiphilic DNA block copolymers. The DNA molecular brushes self-assemble into sub-micron-sized spherical structures in water as evidenced by light scattering as well as atomic force and electron microscopy imaging. The nucleotide sequences remain functional, as assessed by UV and fluorescence spectroscopy subsequent to isoindol synthesis at the surface of the structures. The determination of a vesicular morphology is supported by encapsulation and subsequent spectroscopy monitoring of the release of a water-soluble dye and spectroscopic quantification of the hybridization efficiency (30% in average) of the functional nucleic acid strands engaged in structure formation: about one-half of the nucleotide sequences are available for hybridization, whereas the other half are hindered within the self-assembled structure. Because speciation between complementary and non complementary sequences in the medium could be ascertained by confocal laser scanning microscopy, the stable self-assembled molecular brushes demonstrate the potential for sensing applications.
Herein, we address the question whether anionâÏ and cationâÏ interactions can take place simultaneously on the same aromatic surface. Covalently positioned carboxylateâguanidinium pairs on the surface of 4-amino-1,8-naphthalimides are used as an example to explore pushâpull chromophores as privileged platforms for such âionâpairâÏâ interactions. In antiparallel orientation with respect to the pushâpull dipole, a bathochromic effect is observed. A red shift of 41 nm found in the least polar solvent is in good agreement with the 70 nm expected from theoretical calculations of ground and excited states. Decreasing shifts with solvent polarity, protonation, aggregation, and parallel carboxylateâguanidinium pairs imply that the intramolecular Stark effect from antiparallel ionâpairâÏ interactions exceeds solvatochromic effects by far. Theoretical studies indicate that carboxylateâguanidinium pairs can also interact with the surfaces of Ï-acidic naphthalenediimides and Ï-basic pyrenes.
Direct coupling of carbenium ions with indoles and anilines for the synthesis of cationic π-conjugated dyes
R. Vanel, F.-A. Miannay, E. Vauthey and J. Lacour ChemComm, 50 (81) (2014), p12169-12172
DOI:10.1039/C4CC05193A | unige:40250 | Abstract | Article HTML | Article PDF
A C-C bond forming reaction occurs spontaneously between tris-(2,6-dimethoxyphenyl)carbenium ion and indoles / anilines. The carbocation acts both as electrophile and oxidant. Effective cationic Ï-conjugated dyes are formed resulting in a strong hyper- and bathochromism
Excited-State Dynamics of an Environment-Sensitive Push–Pull Diketopyrrolopyrrole: Major Differences between the Bulk Solution Phase and the Dodecane/Water Interface
S. Richert, S. Mosquera Vazquez, M. Grzybowski, D.T. Gryko, A. Kyrychenko and E. Vauthey Journal of Physical Chemistry B, 118 (33) (2014), p9952-9963
DOI:10.1021/jp506062j | unige:39940 | Abstract | Article HTML | Article PDF
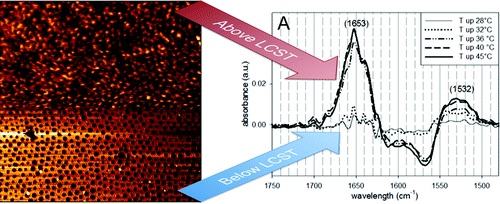
The excited-state dynamics of a diketopyrrolopyrrole (DPP) derivative with pushâpull substituents has been investigated in a variety of solvents and at the dodecane/water and dodecane/heavy-water interfaces using a combination of ultrafast spectroscopic techniques, including transient electronic absorption and time-resolved surface second-harmonic generation. Whereas the photophysics of a nonpolar DPP analogue is mostly independent of the solvent, the fluorescence decay of the pushâpull DPP accelerates strongly by going from aprotic to protic solvents. As this effect increases with the polarity and the hydrogen-bond-donating ability of the solvent, it is attributed to the occurrence of H-bond-assisted nonradiative deactivation induced by the charge-transfer character of the excited state that favors the coupling of the molecule to the H-bond network of the solvent. At the dodecane/water interface, the excited-state lifetime is longer by a factor of ca. 20 than that estimated in pure water and increases further by a factor of about 3 when going to the dodecane/heavy-water interface. This isotope effect, that is more than twice as strong as that measured in bulk solutions, and molecular dynamic simulations indicate that the slowing down of the dynamics at the interface cannot be solely ascribed to a reduced accessibility of the DPP molecule to the aqueous phase. The slower excited-state decay is rather assigned to the conjunction of several effects, such as the strengthening of the H-bond network formed by the interfacial water molecules and the lower local polarity of the interfacial region.
By using a polyelectrolyte layer gold nanoparticles have been assembled onto a Ge internal reflection element. Upon illumination with visible and near infrared light a strong infrared absorption has been observed, which can be traced to intervalence band transitions in Ge. This reveals the existence of holes in the Ge near its valence band edge. The switching between bright and dark states is faster than 160 μs and the device acts as an infrared modulator. The effect develops with a peculiar kinetics, which may indicate the development of an interfacial layer between germanium and gold that allows efficient electron transfer upon illumination.
Light-induced spin-state switching in the mixed crystal series of the 2D coordination network {[Zn1-xFex(bbtr)3](BF4)2}: optical spectroscopy and cooperative effects
P. Chakraborty, C. Enachescu, A. Humair, L. Egger, T. Delgado, A. Tissot, L. Guénée, C. Besnard, R. Bronisz and A. Hauser Dalton Transactions, 43 (47) (2014), p17786-17796
DOI:10.1039/C4DT01728E | unige:42340 | Abstract | Article HTML | Article PDF
Depending on the iron(II) concentration, the mixed crystals of {[Zn1-xFex(bbtr)3](BF4)2}â, bbtr = 1,4-di(1,2,3-triazol-1-yl)butane, 0.01 ⤠x ⤠1, show macroscopic light-induced bistability between the high-spin and the low-spin state. In the highly diluted system with x = 0.01 and up to x = 0.31, the photoinduced low-spin state always relaxes back to the high-spin state independent of the initial light-induced low-spin fraction. In the highly concentrated mixed crystals with x = 0.67, 0.87 and 1, the strong cooperative effects coupled to a crystallographic phase transition result in light-induced bistability with decreasing critical light-induced low-spin fraction and increasing hysteresis width for increasing iron(II) concentrations. The lower limit for the light-induced bistability is estimated at x â 0.5.
The density of atomic systems is analysed via the Single-Exponential Decay Detector (SEDD). SEDD is a scalar field designed to explore mathematical, rather than physical, properties of electron density. Nevertheless, it has been shown that SEDD can serve as a descriptor of bonding patterns in molecules as well as an indicator of atomic shells [P. de Silva, J. Korchowiec, and T. A. Wesolowski, ChemPhysChem13, 3462 (2012)]. In this work, a more detailed analysis of atomic shells is done for atoms in the LiâXe series. Shell populations based on SEDD agree with the Aufbau principle even better than those obtained from the Electron Localization Function, which is a popular indicator of electron localization. A link between SEDD and the local wave vector is given, which provides a physical interpretation of SEDD.
Frozen-Density-Embedding Theory (FDET) is a formalism to obtain the upper bound of the ground-state energy of the total system and the corresponding embedded wavefunction by means of Euler-Lagrange equations [T. A. Wesolowski, Phys. Rev. A77(1), 012504 (2008)]. FDET provides the expression for the embedding potential as a functional of the electron density of the embedded species, electron density of the environment, and the field generated by other charges in the environment. Under certain conditions, FDET leads to the exact ground-state energy and density of the whole system. Following Perdew-Levy theorem on stationary states of the ground-state energy functional, the other-than-ground-state stationary states of the FDET energy functional correspond to excited states. In the present work, we analyze such use of other-than-ground-state embedded wavefunctions obtained in practical calculations, i.e., when the FDET embedding potential is approximated. Three computational approaches based on FDET, that assure self-consistent excitation energy and embedded wavefunction dealing with the issue of orthogonality of embedded wavefunctions for different states in a different manner, are proposed and discussed.
Rather than lead to the usual deoxygenation pathway, metal carbenes derived from α-diazo-β-ketoesters undergo three-atom insertions into epoxides using a combination of 1,10-phenanthroline and [CpRu(CH3CN)3][BArF] as the catalyst. Original 1,4-dioxene motifs are obtained as single regio- and stereoisomers. A perfect syn stereochemistry (retention, e.r. up to 97:3) is observed for the ring opening, which behaves as an SN1-like transformation.
Improved persistent luminescence of CaTiO3:Pr by fluorine substitution and thermochemical treatment
S. Yoon, E.H. Otal, A.E. Maegli, L. Karvonen, S.K. Matam, S.G. Ebbinghaus, B. Walfort, H. Hagemann, S. Pokrant and A. Weidenkaff Journal of Alloys and Compounds, 613 (2014), p338-343 Keywords: CaTiO3:Pr; fluorination; persistent luminescence; afterglow; X-ray diffraction
DOI:10.1016/j.jallcom.2014.06.041 | unige:38546 | Abstract | Article HTML | Article PDF
Fluorine-substituted CaTiO3:Pr phosphors were prepared by a solid-state reaction. Rietveld refinements of powder X-ray diffraction patterns revealed that increasing fluorine-substitution leads to the gradual shrinkage of the unit-cell. Enhanced afterglow intensities were observed with fluorine-substitution. Furthermore, the effect of annealing atmosphere was investigated by thermochemical treatment in different atmospheres (Ar, air and NH3). UV-Vis diffuse reflectance spectra and photoluminescence excitation spectra revealed that Pr4+ in the pristine CaTi(O,F)3:Pr phosphor was partially reduced to Pr3+ under NH3 flow leading to an intensity improvement of ca. 450% compared to CaTiO3:Pr. The substantial improvement of afterglow intensity by fluorine substitution and annealing in NH3 is considered to be connected with the generation of oxygen vacancies and the partial reduction of Pr4+ to Pr3+.
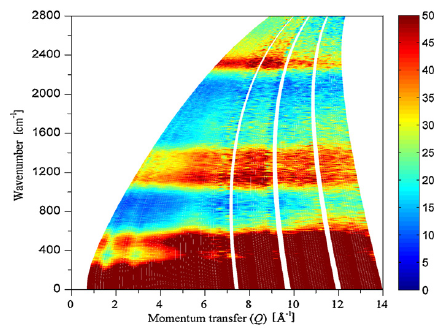
The Raman spectra of a series of monolayer-protected gold clusters were investigated with special emphasis on the AuâS modes below 400 cmâ1. These clusters contain monomeric (SR-Au-SR) and dimeric (SR-Au-SR-Au-SR) goldâthiolate staples in their surface. In particular, the Raman spectra of [Au25(2-PET)18]0/â, Au38(2-PET)24, Au40(2-PET)24, and Au144(2-PET)60 (2-PET = 2-phenylethylthiol) were measured in order to study the influence of the cluster size and therefore the composition with respect to the monomeric and dimeric staples. Additionally, spectra of Au25(2-PET)18â2x(S-/rac-BINAS)x (BINAS = 1,1â²-binaphthyl-2,2â²-dithiol), Au25(CamS)18 (CamS = 1R,4S-camphorthiol), and AunBINASm were measured to identify the influence of the thiolate ligand on the AuâS vibrations. The vibrational spectrum of Au38(SCH3)24 was calculated which allows the assignment of bands to vibrational modes of the different staple motifs. The spectra are sensitive to the size of the cluster and the nature of the ligand. AuâSâC bending around 200 cmâ1 shifts to slightly higher wavenumbers for the dimeric as compared to the monomeric staples. Radial AuâS modes (250â325 cmâ1) seem to be sensitive toward the staple composition and the bulkiness of the ligand, having higher intensities for long staples and shifting to higher wavenumbers for sterically more demanding ligands. The introduction of only one BINAS dithiol has a dramatic influence on the AuâS vibrations because the molecule bridges two staples which changes their vibrational properties completely.
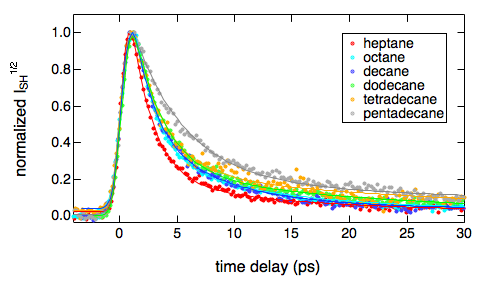
The dynamics of photoinduced electron transfer between polar acceptors and donors has been investigated in apolar solvents using femtosecond-resolved fluorescence spectroscopy. It was found to be ultrafast and to continuously accelerate by varying the excitation wavelength from the maximum to the red edge of the absorption band of the acceptor, the overall difference being as large as a factor 4â5. This violation of the KashaâVavilov rule is explained by a correlation between the composition of the acceptor environment and its transition energy, that is, the more donors around an acceptor, the longer its absorption wavelength, and the faster the quenching. Because of preferential solvation, this dependence is already observed at low quencher concentrations. This effect, which requires quenching to be faster than the fluctuations of the environment composition, should be quite general for photoinduced charge transfer processes in low-polarity, viscous, or rigid media, such as those used in organic optoelectronic devices.
The excited-state dynamics of rhodamine 6G (R6G) has been investigated in aqueous solution using ultrafast transient absorption spectroscopy and at the dodecane/water interface using the femtosecond time-resolved surface second harmonic generation (SSHG) technique. As the R6G concentration exceeds ca. 1 mM in bulk water, both R6G monomers and aggregates are excited to a different extent when using pump pulses at 500 and 530 nm. The excited-state lifetime of the monomers is shortened compared to dilute solutions because of the occurrence of excitation energy transfer to the aggregates, which themselves decay nonradiatively to the ground state with a ca. 70 ps time constant. At the dodecane/water interface, both monomers and aggregates contribute to the SSHG signal to an extent that depends on the bulk concentration, the pump and probe wavelengths, and the polarization of probe and signal beams. The excited-state lifetime of the monomers at the interface is of the order of a few picoseconds even at bulk concentrations where it is as large as several nanoseconds. This is explained by the relatively high interfacial affinity of R6G that leads to a large interfacial concentration, favoring aggregation and thus rapid excitation energy transfer from monomers to aggregates.
Inorganic compounds with BH4-Â ions are the subject of many recent investigations in the context of potential hydrogen storage materials. In this work, Attenuated Total Reflectance Fourier Transform Infrared (ATR-FTIR) spectra of a series of reference and research compounds (including deuterated samples) are collected and made available to the research community.
Anisotropic magnetic, transport and thermodynamic properties of novel tetragonal Ce2RhGa12 compound
S. Nallamuthu, T.P. Rashid, V. Krishnakumar, C. Besnard, H. Hagemann, M. Reiffers and R. Nagalakshmi Journal of Alloys and Compounds, 604 (2014), p379-383 Keywords: intermetallic; anisotropy; heavy fermions; heat capacity; magnetization
DOI:10.1016/j.jallcom.2014.03.067 | unige:37941 | Article HTML
We report on a comprehensive study of the magnetization, resistivity and heat capacity on the single crystals of Ce2RhGa12 synthesized using Ga flux. Single crystal X-ray diffraction data confirm the tetragonal Pb/nbm structure of Ce2RhGa12, which is isostructural to Ce2PdGa12. Ce2RhGa12 orders antiferromagnetically at TN = 3.5 K and exhibits anisotropic magnetic behavior, inferred from the magnetization and resistivity data, taken along the two principal crystallographic directions of the crystal, viz., along [100] and [001]. The anisotropic magnetic response of Ce2RhGa12 establishes [001] as the easy axis of magnetization, and a weak meta-magnetic transition is also observed in the magnetic isotherm at 2K along the same axis. A sharp peak in specific heat signals the bulk antiferromagnetic transition at TN = 3.5 K, which shifts to lower temperatures in low applied fields. The electrical resistivity along the two directions shows metallic behavior from 300K down to 1.8K and establishes Ce2RhGa12 as a normal, trivalent cerium compound.
The role of ligand-field states in the ultrafast photophysical cycle of the prototypical iron(II) spin-crossover compound [Fe(ptz)6](BF4)2
A. Marino, P. Chakraborty, M. Servol, M. Lorenc, E. Collet and A. Hauser Angewandte Chemie International Edition, 53 (15) (2014), p3863-3867 Keywords: intersystem crossing;LIESST;ligand-field states;spin crossover;ultrafast spectroscopy
DOI:10.1002/anie.201310884 | unige:38562 | Abstract | Article HTML | Article PDF
Light-induced excited spin state trapping (LIESST) in iron(II) spin-crossover compounds, i.e., the light-induced population of the high-spin (S=2) state below the thermal transition temperature, was discovered thirty years ago. For irradiation into metal-ligand charge transfer (MLCT) bands of the low-spin (S=0) species the acknowledged sequence takes the system from the initially excited 1MLCT to the high-spin state via the 3MLCT state within ~150 fs, thereby bypassing low-lying ligand-field (LF) states. Nevertheless, these play role, as borne out by the observation of LIESST and reverse-LIESST on irradiation directly into the LF bands for systems with only high-energy MLCT states. Herein we elucidate the ultrafast reverse-LIESST pathway by identifying the lowest energy S=1 LF state as intermediate state with a lifetime of 39 ps for the light-induced high-spin to low-spin conversion on irradiation into the spin-allowed LF transition of the high-spin species in the NIR.
The dynamics of bimolecular photoinduced electron-transfer reactions has been investigated with three donor/acceptor (D/A) pairs in tetrahydrofuran (THF) and acetonitrile (ACN) using a combination of ultrafast spectroscopic techniques, including time-resolved infrared absorption. For the D/A pairs with the highest driving force of electron transfer, all transient spectroscopic features can be unambiguously assigned to the excited reactant and the ionic products. For the pair with the lowest driving force, three additional transient infrared bands, more intense in THF than in ACN, with a time dependence that differs from those of the other bands are observed. From their frequency and solvent dependence, these bands can be assigned to an exciplex. Moreover, polarization-resolved measurements point to a relatively well-defined mutual orientation of the constituents and to a slower reorientational time compared to those of the individual reactants. Thanks to the minimal overlap of the infrared signature of all transient species in THF, a detailed reaction scheme including the relevant kinetic and thermodynamic parameters could be deduced for this pair. This analysis reveals that the formation and recombination of the ion pair occur almost exclusively via the exciplex.
Probing the Anisotropic Distortion of Photoexcited Spin Crossover Complexes with Picosecond X‑ray Absorption Spectroscopy
S.E. Canton, X. Zhang, L.M. Lawson Daku, A.L. Smeigh, J. Zhang, Y. Liu, C.-J. Wallentin, K. Attenkofer, G. Jennings, C.A. Kurtz, D. Gosztola, K. Wärnmark, A. Hauser and V. Sundström Journal of Physical Chemistry C, 118 (8) (2014), p4536-4545
DOI:10.1021/jp5003963 | unige:37983 | Abstract | Article HTML | Article PDF
For numerous spin crossover complexes, the anisotropic distortion of the first coordination shell around the transition metal center governs the dynamics of the high-spin/lowspin interconversion. However, this structural parameter remains elusive for samples that cannot be investigated with crystallography. The present work demonstrates how picosecond X-ray absorption spectroscopy is able to capture this specifi c deformation in the photoinduced high-spin state of solvated [Fe(terpy)2 ]2+ , a complex which belongs to the prominent family of spin crossover building blocks with nonequivalent metalâ ligand bonds. The correlated changes in FeâNAxial , Feâ NDistal , and bite angle NDistalâ Feâ NAxial extracted from the measurements are in very good agreement with those predicted by DFT calculations in D2d symmetry. The outlined methodology is generally applicable to the characterization of ultrafast nuclear rearrangements around metal centers in photoactive molecular complexes and nanomaterials, including those that do not display long-range order.
The excited-state dynamics of two multichromophoric arrays composed of a naphthalene diimide centre and four zinc or free-base porphyrins substituted on the naphthalene core via aniline bridges has been investigated using a combination of stationary and ultrafast spectroscopies. These pentads act as efficient antennae as they absorb over the whole visible region, with a band around 700 nm, associated with a transition to the S1 state delocalised over the whole arrays, and bands at higher energy due to transitions centred on the porphyrins. In non-polar solvents, population of these porphyrin states is followed by sub-picosecond internal conversion to the S1 state. The existence of a charge-separated state located above the S1 state could enhance this process. The decay of the S1 state is dominated by non-radiative deactivation on the 100 ps timescale, most probably favoured by the small S1-S0 energy gap and the very high density of vibrational states of these very large chromophores. In polar solvents, the charge-separated state lies just below the S1 state. It can be populated within a few picoseconds by a thermally-activated hole transfer from the S1 state as well as via sub-picosecond non-equilibrium electron transfer from vibrationally hot porphyrin excited states. Because of the small energy gap between the charge-separated state and the ground state, charge recombination is almost barrierless and occurs within a few picoseconds. Despite their very different driving forces, charge separation and recombination occur on similar timescales. This is explained by the electronic coupling that differs considerably for both processes.
Retinal is the light-absorbing biochromophore responsible for the activation of vision pigments and light-driven ion-pumps. Nature has evolved molecular tuning mechanisms that significantly shift the optical properties of the retinal pigments to enable their absorption of visible light. Using large-scale quantum chemical calculations at the density functional theory level combined with the frozen density embedding theory approach, we show here how the protein environment of vision pigments tune the absorption of retinal to the 2.3-2.6 eV (480-530 nm) region by electrostatically dominated interactions between the chromophore and the surrounding protein residues. The calculations accurately reproduce the experimental absorption maxima of rhodopsin (2.49 eV/498 nm), and the red, green, and blue color pigments (2.3-2.9 eV/430-530 nm). We further identify key interactions responsible for the red- and blue-shifting effects by mutating the rhodopsin structure in silico, and find that deprotonation of the retinyl is likely to be responsible for the blue shifted absorption in the blue cone vision pigment.
Oxadiazole based bipolar host materials employing planarized triarylamine donors for RGB PHOLEDs with low efficiency roll-off
P. Kautny, D. Lumpi, Y. Wang, A. Tissot, J. Bintinger, E. Horkel, B. Stoeger, C. Hametner, H. Hagemann, D. Ma and J. Fröhlich Journal of Materials Chemistry C, 2 (11) (2014), p2069-2081
DOI:10.1039/c3tc32338b | unige:34399 | Abstract | Article HTML | Article PDF
A series of 6 novel triarylamine-containing oxadiazole compounds (o-PCzPOXD, o-ICzPOXD, o-TPATOXD, o-PCzTOXD, o-ICzTOXD, o-CzTOXD) have been designed, synthesized and characterized concerning applications as host materials in PHOLED devices. To further improve the ortho-linkage concept, the impact of incorporating planarized electron-donating triarylamine (TAA) structures on intramolecular charge transfer was examined. The effect was evaluated for two series of electron-accepting oxadiazole scaffolds, realizing ortho-linkage on the benzene (POXD) and the thiophene (TOXD) core. Thermal analysis shows increased glass-transition temperatures for planarized structures indicating an improved morphological stability. A higher degree of planarization also results in significantly increased singlet and triplet energy values, revealing the impact on the intramolecular charge transfer. Employing the developed materials, red (o-TPATOXD: CEmax: 28.8 cd A-1, EQEmax: 16.9%), green (o-PCzPOXD: CEmax: 62.9 cd A-1, EQEmax: 17.1%) and blue (o-PCzPOXD: CEmax: 29.8 cd A-1, EQEmax: 13.4%) devices were achieved showing remarkably low efficiency roll-off for planarized donors. Hence, this is the first report of efficient blue devices for this specific class of host materials. It is proposed that the results correlate with an increasing ortho-linkage effect and decreasing donor strength of the TAA moiety by planarization and, thus, tackling one of the major challenges in PHOLED research: improving both triplet energy and compound stability.
To access the intrinsic, diffusion free, rate constant of bimolecular photoinduced electron transfer reactions, fluorescence quenching experiments have been performed with 14 donor/acceptor pairs, covering a driving-force range going from 0.6 to 2.4 eV, using steady-state and femtosecond time-resolved emission, and applying a diffusion-reaction model that accounts for the static and transient stages of the quenching for the analysis. The intrinsic electron transfer rate constants are up to 2 orders of magnitude larger than the diffusion rate constant in acetonitrile. Above ~1.5 eV, a slight decrease of the rate constant is observed, pointing to a much weaker Marcus inverted region than those reported for other types of electron transfer reactions, such as charge recombination. Despite this, the driving force dependence can be rationalized in terms of Marcus theory.
Vapor pressure measurements of Mg(BH4)2 using Knudsen torsion effusion thermo graphic method
L.-N.N. Nforbi, A. Talekar, K.H. Lau, R. Chellapa, W.-M. Chien, D. Chandra, H. Hagemann, Y. Filinchuk, J.-C. Zhao and A. Levchenko International Journal of Hydrogen Energy, 39 (5) (2014), p2175-2186 Keywords: Mg(BH4)2; hydrogen desorption under dynamic vacuum; torsion effusion vapor pressure measurements; vaporization thermodynamics
DOI:10.1016/j.ijhydene.2013.11.071 | unige:33261 | Abstract | Article PDF
The vapor pressure and molecular weight of effusing vapors of α, β, and amorphous Mg(BH4)2 were determined by Torsion-effusion gravimetric method, under dynamic vacuum. A Cahn balance in the system yielded the rate of the weight loss. Molecular weights measured revealed if the effusion was congruent or there was disproportionation. The vaporization behavior of crystalline Mg(BH4)2, was measured up to 533 K at pressures of â¼10â5 torr. It was found that Mg(BH4)2 disproportionates to form predominantly H2 gas (â¼95%) with a small amount of Mg(BH4)2 (â¼5%) in the gas phase. The combined average molecular weight measured is 4.16 g/mol. The equations for vapor pressures for crystalline Mg(BH4)2 are given by: log PTotal(bar) = 9.2303 â 7286.2/T, log PMg(BH4)2 (bar) = 8.2515 - 7286.2 / T , and log PH2 (bar) = 9.1821 - 7286.2 / T. The partial pressures of the gaseous species were determined as PMg2(4BH)(g)/PT=0.105 and PH2(g)/PT=0.895. Enthalpies of vaporization for the effusing gases were calculated to be ÎH = +558.0 kJ/mol H2 and ÎH = +135 kJ/mol Mg(BH4)2. The standard Gibbs free energy changes, ÎG°(kJ/mol), for the complete decomposition reaction (Mg(BH4)2(s) â Mg(s) + 2B(s) + 4H2(g)), sublimation reaction (Mg(BH4)2(s) â Mg(BH4)2(g)) and the disproportionation reaction for Mg(BH4)2 are reported in this paper. The decomposition pathway of amorphous Mg(BH4)2 was also carried out between 388.2 K and 712.8 K showing multistep decomposition of a-Mg(BH4)2 Different reaction products were obtained depending on the method used in the vaporization experiment. The behavior of the amorphous Mg(BH4)2(s) is very different from those for the two crystalline phases (α and β). The vapor pressure behavior and thermodynamics of vaporization of different phases of Mg(BH4)2 are presented.
How to choose the frozen density in Frozen-Density Embedding Theory-based numerical simulations of local excitations?
M. Humbert-Droz, X. Zhou, S.V. Shedge and T.A. Wesolowski Theoretical Chemistry Accounts, 133 (1) (2014), p1405 Keywords: frozen-density; embedding theory; linear-response; time-dependent density functional
DOI:10.1007/s00214-013-1405-1 | unige:33263 | Abstract | Article HTML | Article PDF
According to Frozen-Density Embedding Theory, any observable evaluated for the embedded species is a functional of the frozen density (ÏB âthe density associated with the environment). The environment-induced shifts in the energies of local excitations in organic chromophores embedded in hydrogen-bonded environments are analyzed. The excitation energies obtained for ÏB , which is derived from ground-state calculations for the whole environment applying medium quality basis sets (STOâDZP) or larger, vary in a narrow range (about 0.02 eV which is at least one order of magnitude less than the magnitude of the shift). At the same time, the ground-state dipole moment of the environment varies significantly. The lack of correlation between the calculated shift and the dipole moment of the environment reflects the fact that, in Frozen-Density Embedding Theory, the partitioning of the total density is not unique. As a consequence, such concepts as âenvironment polarizationâ are not well defined within Frozen-Density Embedding Theory. Other strategies to generate ÏB (superposition of densities of atoms/molecules in the environment) are shown to be less robust for simulating excitation energy shifts for chromophores in environments comprising hydrogen-bonded molecules.
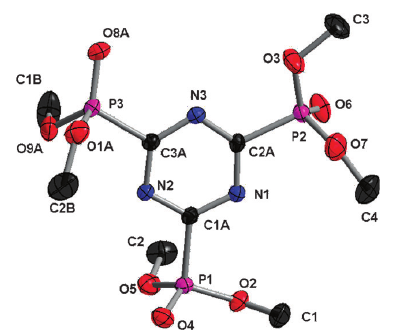
A large pi-conjugated chromophore composed of two dipyrido[3,2-a:2â,3â-c]phenazine (dppz) units directly fused to the central tetrathiafulvalene (TTF) core, has been prepared as a bridging ligand, and its strong binding ability to Ru2+ forming a new dinuclear complex is presented. The electronic absorption and luminescence and the electrochemical behaviour of the free ligand as well as the Ru2+ complex have been investigated in detail. The free ligand shows a very strong band in the UV region consistent with ligand centred Ï-Ï* transitions and an intense broad band in the visible region corresponding to an intramolecular charge transfer (ILCT) transition. Upon coordination, a metal-to-ligand charge transfer (MLCT) appears at 22520 cm-1 while the ILCT band is bathochromically shifted by 1620 cm-1. These electrochemically amphoteric chromophores have also been characterized by spectroelectrochemical methods. The oxidized radical species of the free ligand show a strong tendency to undergo aggregation, in which long-distance attractive interactions overcome the electrostatic repulsion. Moreover, these two new chromophores reveal an ILCT fluorescence with large solvent-dependent Stokes shifts and quantum efficiencies of 0.052 for the free ligand and 0.016 for its dinuclear Ru2+ complex in CH2Cl2.
Discriminability of tryptophan containing dipeptides using quantum control
S. Afonina, O. Nenadl, A. Rondi, L. Bonacina, J. Extermann, D. Kiselev, I. Dolamic, T. Burgi and J.P. Wolf Applied Physics B, 111 (4) (2013), p541-549
DOI:10.1007/s00340-013-5370-0 | unige:37054 | Abstract | Article HTML | Article PDF
We show that the coherent manipulation of molecular wavepackets in the excited states of trp-containing dipeptides allows efficient discrimination among them. Optimal dynamic discrimination fails, however, for some dipeptide couples. When considering the limited spectral resources at play (3 nm bandwidth at 266 nm), we discuss the concept of discriminability, which appears uncorrelated to both static spectra and relaxation lifetimes.
Model-free Investigation of Ultrafast Bimolecular Chemical Reactions: Bimolecular Photo Induced Electron Transfer
B. Lang, A. Rosspeintner and E. Vauthey EPJ Web of Conferences, 41 (2013), p5041
DOI:10.1051/epjconf/20134105041 | unige:94113 | Abstract | Article PDF
Using photoinduced bimolecular electron transfer reactions as example we demonstrate how diffusion controlled bimolecular chemical reactions can be studied in a model-free manner by quantitatively combining different ultrafast spectroscopical tools.
Attenuated total reflection infrared (ATR-IR) spectroscopy is used to study the adsorption of gold and silver nanoparticles and the layer-by-layer (LBL) growth of polyelectrolyte multilayers on a Ge ATR crystal. The Ge ATR crystal is first functionalized using positively charged polyelectrolyte poly(allylamine hydrochloride) (PAH). Then citrate-stabilized gold or silver nanoparticles are adsorbed onto the modified Ge ATR crystal. When gold or silver nanoparticles are adsorbed, a drastic increase of the water signal is observed which is attributed to an enhanced absorption of IR radiation near the nanoparticles. This enhancement was much larger for the silver nanoparticles (SNP). On top of the nanoparticles multilayers of oppositely charged polyelectrolytes PAH and poly(sodium 4-styrenesulfonate) (PSS) were deposited, which allowed to study the enhancement of the IR signals as a function of the distance from the nanoparticles. Furthermore, adsorption of a thiol, N-acetyl-l-cysteine, on the nanoparticles confirmed the enhancement. In the case of SNP an absorbance signal of about 15% was observed, which is a factor of about 40 times larger compared to typical signals measure without nanoparticles.
Modeling Transition Metal Complexes in the Frameworkof the Spin-Crossover Phenomenon: a DFT Perspective
L.M. Lawson Daku Current Inorganic Chemistry, 3 (3) (2013), p242-259 Keywords: density functional theory, spin crossover, transition metal complexes
DOI:10.2174/1877944103666140110231029 | unige:73072
Using the study of the low-spin complex [Fe(bpy)3]2+ in the gas phase and in condensed phases as a guideline, we examine different aspects of the application of DFT to the study of transition metal complexes in the framework of spin crossover or related phenomena.
Crystal structure solution of an elusive polymorph of Dibenzylsquaramide
A. Portell, X. Alcobé, L.M. Lawson Daku, R. Cerny and R. Prohens Powder Diffraction, 28 (S2) (2013), p470-480 Keywords: dibenzylsquaramide; crystal structure; X-ray powder diffraction
DOI:10.1017/S0885715613000821 | unige:35159 | Abstract | Article PDF
The crystal structure of the third polymorph of dibenzylsquaramide (Portell, A. et al., 2009), (fig. 1) has been determined from laboratory X-ray powder diffraction data by means of direct space methods using the computing program FOX. (Favre-Nicolin and Äerný, 2002) The structure resolution has not been straightforward due to several difficulties on the indexing process and in the space group assignment. The asymmetric unit contains two different conformers, which has implied an additional difficulty during the Rietveld (Rietveld, 1969) refinement. All these issues together with particular structural features of disquaramides are discussed.
Light-upconversion via stepwise energy transfer from a sensitizer to an activator exploits linear optics for converting low-energy infrared or near-infrared incident photons to higher energy emission occurring in the part of the electromagnetic spectrum ranging from visible to ultraviolet. Stepwise excitation is restricted to activators possessing intermediate long-lived excited states such as those found for trivalent lanthanide cations dispersed in solid-state matrices. When the activator is embedded in a molecular complex, efficient non-radiative relaxation processes usually reduce excited state lifetimes to such an extent that upconversion becomes too inefficient to be detected under practical excitation intensities. Theoretical considerations suggest that the combination of millisecond timescale sensitizers with a central lanthanide activator located in supramolecular complexes circumvents this bottleneck by creating a novel pathway reminiscent of the energy transfer upconversion mechanism observed in doped solids. Application of this novel concept to chromium/erbium pairs in discrete triple-stranded helicates demonstrates that strong-field trivalent chromium chromophores irradiated with near-infrared photons produce upconverted green erbium-centered emission both in the solid state and in solution.
A Pt(II) complex with both a phenanthroline and a tetrathiafulvalene-extended dithiolate ligand: Synthesis, crystal structure, electro-chemical and spectroscopic properties
C. Jia, J. Ding, S.-X. Liu, G. Labat, A. Neels, A. Hauser and S. Decurtins Polyhedron, 55 (2013), p87-91 Keywords: tetrathiafulvalene; intramolecular charge-transfer; spectroelectrochemistry
DOI:10.1016/j.poly.2013.02.064 | unige:32100 | Article HTML | Article PDF
The reaction of 4,5-bis(2'-cyano-ethylsulfanyl)-4',5'-dipropylthiotetrathiafulvalene with [Pt(phen)Cl2] (phen = 1,10-phenanthroline) with CsOH as base in CH3OHâTHF affords the target complex 1 in 44% yield. This complex crystallizes in the monoclinic space group P21/c, M = 790.01, a = 12.1732(12), b = 15.851(2), c = 14.5371(16) Ã , b = 107.693(12)Ë, V = 2672.4(5) Ã 3 and Z = 4. It undergoes two reversible single-electron oxidation and two irreversible reduction processes. An intense electronic absorption band at 15200 cm-1 (658 nm) in CH2Cl2 is assigned to the intramolecular mixed metal/ligand-to-ligand charge transfer (LLCT) from a tetrathiafulvalene-extended dithiolate-based HOMO to a phenanthroline-based LUMO. This band shifts hypsochromically with increasing solvent polarity. Systematic changes in the optical spectra upon oxidation allow precise tuning of the oxidation states of 1 and reversible control over its optical properties. Irradiation of 1 at 15625 cm-1 (640 nm) in glassy solution below 150 K results in emission from the 3LLCT excited state.
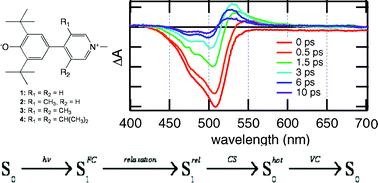
The excited-state dynamics of two donorâacceptor biaryls that differ by the strength of the acceptor, a pyridinium or a pyrylium moiety, have been investigated using a combination of steady-state solvatochromic absorption, ultrafast fluorescence, as well as visible and infrared transient absorption spectroscopies. The negative solvatochromic behavior of pyridinium phenolate indicates that the permanent electric dipole moment experiences a decrease upon S1 â S0 excitation, implying that the ground state possesses more zwitterionic character than the excited state. In contrast, pyrylium phenolate exhibits a weakly positive solvatochromic behavior corresponding to a small increase in the dipole moment upon excitation, implying more zwitterionic character in the excited than the ground state. Both compounds are therefore situated at different sides of the cyanine-limit structure, which has equally polar ground and excited states. Despite these differences, both molecules exhibit qualitatively similar excited-state properties. They are characterized by a very short fluorescence lifetime, increasing from about 1 to 20 ps, when varying solvent viscosity from 0.4 to 11 cP. There are, however, characteristic differences between the two compounds: The excited-state lifetimes of the pyrylium dye are shorter and also depend somewhat on polarity. The ensemble of spectroscopic data can be explained with a model where the emitting FranckâCondon excited state relaxes upon twisting around the single bond between the aryl units to a point where the excited- and ground-state surfaces are very close or intersect. After internal conversion to the ground state, the distorted molecule relaxes back to its equilibrium planar configuration, again largely dependent upon solvent viscosity. However, in this case, the kinetics for the pyrylium dye are slower than for the pyridinium dye and the polar solvent-induced acceleration is significantly stronger than in the excited state. This difference of kinetic behavior between the two compounds is a direct consequence of the change of the electronic structure from anormal to an overcritical merocyanine evidenced by steady-state spectroscopy.
The excited-state dynamics of the cationic dye malachite green (MG) and of the dianionic dye eosin B at the dodecane/water interface has been investigated using femtosecond time-resolved surface second harmonic generation (TR-SSHG). By using different probe wavelengths, the contributions of monomeric and aggregated MG to the signal could be spectroscopically distinguished. The effect of the addition of a small amount of surfactants was found to strongly depend on the relative charges of surfactant and dye. For surfactant/dye pairs with opposite charges, the TR-SSHG signal is dominated by the contribution from aggregates, whereas for pairs with the same charges, the signal intensity becomes vanishingly small. These effects are explained in terms of electrostatic interactions between surfactants and dyes that favor either attraction of the dye toward the interface or its repulsion toward the bulk. As a very similar behavior is observed with MG upon addition of NaSCN, we conclude that, in this case, this effect reflects the affinity of SCN¯ for the interface. On the other hand, the guanidinium cation was found to have a different effect than that of a positively charged surfactant on the SSHG signal of MG, indicating this cation does not accumulate in the interfacial region.
Acylgermanes: Photoinitiators and Sources for Ge-Centered Radicals. Insights into their Reactivity
D. Neshchadin, A. Rosspeintner, M. Griesser, B. Lang, S. Mosquera-Vazquez, E. Vauthey, V. Gorelik, R. Liska, C. Hametner, B. Ganster, R. Saf, N. Moszner and G. Gescheidt Journal of the American Chemical Society, 135 (46) (2013), p17314-17321
DOI:10.1021/ja404433u | unige:31278 | Abstract | Article HTML | Article PDF
Acylgermanes have been shown to act as efficient photoinitiators. In this investigation we show how dibenzoyldiethylgermane 1 reacts upon photoexcitation. Our real-time investigation utilizes femto- and nanosecond transient absorption, time-resolved EPR (50 ns), photo-chemically induced dynamic nuclear polarization, DFT calculations, and GC-MS analysis. The benzoyldiethylgermyl radical G⢠is formed via the triplet state of parent 1. On the nanosecond time scale this radical can recombine or undergo hydrogen-transfer reactions. Radical G⢠reacts with butyl acrylate at a rate of 1.2 ± 0.1 à 108 and 3.2 ± 0.2 à 108 Mâ1 sâ1, in toluene and acetonitrile, respectively. This is Ë1 order of magnitude faster than related phosphorus-based radicals. The initial germyl and benzoyl radicals undergo follow-up reactions leading to oligomers comprising GeâO bonds. LC-NMR analysis of photocured mixtures containing 1 and the sterically hindered acrylate 3,3-dimethyl-2-methylenebutanoate reveals that the products formed in the course of a polymerization are consistent with the intermediates established at short time scales.
The far infrared spectra of a series of well-defined gold clusters covered by 2-phenylethanetiolate were studied. The spectra of the cluster are different but the differences are subtle. The Au-S stretching vibrations give rise to bands around 300 cm-1 and below. The relative intensity of these bands changes but they shift only slightly for different clusters. A low-frequency band was identified that is sensitive to the conformation (trans / gauche) of the 2-phenylethanetiolate ligand.
We describe the experimental investigation of time-resolved magnetic field effects in exciplex-forming organic donorâacceptor systems. In these systems, the photoexcited acceptor state is predominantly deactivated by bimolecular electron transfer reactions (yielding radical ion pairs) or by direct exciplex formation. The delayed fluorescence emitted by the exciplex is magnetosensitive if the reaction pathway involves loose radical ion pair states. This magnetic field effect results from the coherent interconversion between the electronic singlet and triplet radical ion pair states as described by the radical pair mechanism. By monitoring the changes in the exciplex luminescence intensity when applying external magnetic fields, details of the reaction mechanism can be elucidated. In this work we present results obtained with the fluorophore-quencher pair 9,10-dimethylanthracene/N,N-dimethylaniline (DMA) in solvents of systematically varied permittivity. A simple theoretical model is introduced that allows discriminating the initial state of quenching, viz., the loose ion pair and the exciplex, based on the time-resolved magnetic field effect. The approach is validated by applying it to the isotopologous fluorophore-quencher pairs pyrene/DMA and pyrene-d10/DMA. We detect that both the exciplex and the radical ion pair are formed during the initial quenching stage. Upon increasing the solvent polarity, the relative importance of the distant electron transfer quenching increases. However, even in comparably polar media, the exciplex pathway remains remarkably significant. We discuss our results in relation to recent findings on the involvement of exciplexes in photoinduced electron transfer reactions.
The ligand exchange reaction between Au38(2-PET)24 (2-PET: 2-phenylethanethiolate) clusters and enantiopure planar chiral [2.2]paracyclophane-4-thiol 1 (PCP-4-SH) was studied using High Performance Liquid Chromatography (HPLC) and mass spectrometry. It is shown that even at the initial stage of the reaction at least three out of the four symmetry-unique sites are exchanged leading to different regioisomers of composition Au38(2-PET)23(PCP-4-S)1. Using HPLC it was possible to isolate one specific regioisomer. The latter is stable at room temperature and at slightly elevated temperatures. However, at 80° C the adsorbed thiolate (PCP-4-S) moves between different symmetry-unique sites. These observations have implications for the preparation of mixed ligand shell clusters with specific ligand patterns.
Molecular Dynamics Simulations of Liquid Phase Interfaces: Understanding the Structure of the Glycerol/Water–Dodecane System
F.R. Beierlein, A.M. Krause, C.M. Jäger, P. Fita, E. Vauthey and T. Clark Langmuir, 29 (38) (2013), p11898-11907
DOI:10.1021/la4021355 | unige:30152 | Abstract | Article HTML | Article PDF
Modern spectroscopic techniques such as time-resolved second-harmonic-generation spectroscopy allow molecules to be examined selectively directly at phase interfaces. Two-phase systems formed by glycerol/water and alkane layers have previously been studied by time-resolved second-harmonic-generation spectroscopic measurements. In this molecular dynamics study, a triphenylmethane dye was inserted at the glycerol/waterâalkane interface and was used as a probe for local properties such as viscosity. We now show how extensive simulations over a wide range of concentrations can be used to obtain a detailed view of the molecular structure at the glycerol/waterâalkane interface. Glycerol is accumulated in a double layer adjacent to the alkane interface, which results in increased viscosity of the glycerol/water phase in the direct vicinity of the interface. We also show that conformational ensembles created by classical molecular-dynamics simulations can serve as input for QM/MM calculations, yielding further information such as transition dipoles, which can be compared with spectroscopic measurements.
Structures and Chiroptical Properties of the BINAS-monosubstituted Au38(SCH3)24 cluster
B. Molina, A. Sánchez-Castillo, S. Knoppe, I.L Garzon, T. Bürgi and A. Tlahuice-Flores Nanoscale, 5 (22) (2013), p10956-10962
DOI:10.1039/c3nr03403h | unige:31281 | Abstract | Article HTML | Article PDF
The structure and optical properties of a set of R-1,1´-binaphthyl-2,2´-dithiol (R-BINAS) monosubstituted A-Au38(SCH3)24 clusters are studied by means of time dependent density functional theory (TD-DFT). While it was proposed earlier that BINAS selectively binds to monomer motifs (SR-Au-SR) covering the Au23 core, our calculations suggest a binding mode that bridges two dimer (SR-Au-SR-Au-RS) motifs. The more stable isomers show a negligible distortion induced by BINAS adsorption on the Au38(SCH3)24 cluster which is reflected by similar optical and Circular Dichroism (CD) spectra to those found for the parent cluster. The results furthermore show that BINAS adsorption does not enhance the CD signals of the Au38(SCH3)24 cluster.
The recently reported crystal structure of the Au28(TBBT)20 cluster (TBBT: para-tert-butylbenzenethiolate) is analyzed with (Time-Dependent-) Density Functional Theory (TD-DFT). Bader charge analysis reveals a novel trimeric Au3(SR)4 binding motif. The cluster can be formulated as Au14(Au2(SR)3)4(Au3(SR)4)2. The electronic structure of the Au146+ core and the ligand-protected cluster were analyzed and their stability can be explained by formation of distorted eight-electron superatoms. Optical absorption and Circular Dichroism (CD) spectra were calculated and compared to the experiment. Assignment of handedness of the intrinsically chiral cluster is possible.
Ultrafast transient absorption spectroscopy serves to identify the 3dd state as intermediate quencher state of the 3MLCT luminescence in the non-luminescent ruthenium complexes [Ru(m-bpy)3]2+ (m-bpy = 6-methyl-2,2â²-bipyridine) and [Ru(tm-bpy)3]2+ (tm-bpy = 4,4â²,6,6â²-tetramethyl-2â²,2â²-bipyridine). For [Ru(m-bpy)3]2+, the population of the 3dd state from the 3MLCT state occurs within 1.6 ps, while the return to the ground state takes 450 ps. For [Ru(tm-bpy)3]2+, the corresponding values are 0.16 and 7.5 ps, respectively. According to DFT calculations, methyl groups added in the 6 and 6â² positions of bipyridine stabilize the 3dd state by â¼4000 cmâ1 each, compared to [Ru(bpy)3]2+.
Hydrogen-fluorine exchange in NaBH4-NaBF4
L. Rude, U. Filso, V. D'Anna, A. Spyratou Stratmann, B. Richter, S. Hino, O. Zavorotynska, M. Baricco, M.H. Sørby, B.C. Hauback, H. Hagemann, F. Besenbacher, J. Skibsted and T.R. Jensen Physical Chemistry Chemical Physics, 15 (2013), p18185-18194
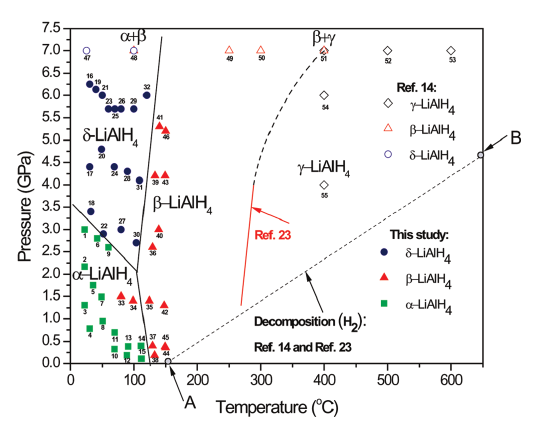
DOI:10.1039/c3cp52815d | unige:30153 | Abstract | Article PDF
Hydrogen-fluorine exchange in the NaBH4âNaBF4 system is investigated with a range of experimental methods combined with DFT calculations and a possible mechanism for the reactions is proposed. Fluorine substitution is observed by in-situ synchrotron radiation powder X-ray diffraction (SR-PXD) as a new Rock salt type compound with idealized composition NaBF2H2 in the temperature range T = 200 to 215 °C. Combined use of solid-state 19F MAS NMR, FT-IR and DFT calculations supports the formation of a BF2H2â complex ion, reproducing the observation of a 19F chemical shift at 144.2 ppm, which is different from that of NaBF4 at 159.2 ppm, along with the new absorption bands observed in the IR spectra. After further heating, the fluorine substituted compound becomes X-ray amorphous and decomposes to NaF at ~310 ºC. This work shows that fluorine-substituted borohydrides tend to decompose to more stable compounds, e.g. NaF, BF3 or amorphous products such as closo-boranes, e.g. Na2B12H12. The NaBH4-NaBF4 composite decomposes at lower temperatures (300 °C) compared to NaBH4 (476 °C), as observed by thermogravimetric analysis. NaBH4-NaBF4 (1:0.5) preserves 30 % of the hydrogen storage capacity after three hydrogen release and uptake cycles compared to 8 % for NaBH4 measured by the Sievertâs method under identical conditions, but more than 50 % using prolonged hydrogen absorption time. The reversible hydrogen storage capacity tends to decrease possibly due to the formation of NaF and Na2B12H12. On the other hand, the additive sodium fluoride appears to facilitate hydrogen uptake, prevent foaming, phase segregation and loss of material from the sample container for samples of NaBH4-NaF.
A major problem in the extraction of the reaction probability in bimolecular processes is the disentanglement from the influence of molecular diffusion. One of the strategies to overcome it makes use of reactive solvents in which the reactants do not need to diffuse to encounter each other. However, most of our quantitative understanding of chemical reactions in solution between free partners is based on the assumption that they can be approximated by spheres because rotation averages their mutual orientations. This condition may not be fulfilled when the reaction takes place on time scales faster than that of molecular reorientation. In this work, the fluorescence quenching of two very similar polyaromatic hydrocarbons with different electric dipole moments is measured. The concentration of a liquid electron-donating quencher is varied from very dilute solutions to pure quencher solutions. In both cases, the thermodynamics of the reactions are very similar and, according to the Marcus expression, the kinetics are expected to proceed at similar rates. However, one of them is 10 times faster in the pure quencher solution. This difference starts at relatively low quencher concentrations. An explanation based on the fluorophoreâsolvent dipoleâdipole interaction and the consequent orientational solvent structure is provided. The orientational correlation between fluorophore and quencher is calculated by means of computer simulations. Important differences depending on the fluorophore dipole moment are found. The kinetics can be explained quantitatively with a reactionâdiffusion model that incorporates the effects of the presence of the dipole moment and the rotational diffusion, only in the highest quencher concentration case, but not in dilute solutions, most likely due to fundamental limitations of the kinetic theory.
The ligand exchange reaction between monodisperse Au25(2-PET)18 (2-PET: 2-phenylethylthiolate) clusters and 1,1â²-binaphthyl-2,2â²-dithiol (BINAS) was long thought to induce decomposition of the cluster (Si et al., J. Phys. Chem. C, 2009). We repeated the experiment and analyzed the reaction products using MALDI-TOF mass spectrometry. The spectra clearly indicate successful ligand exchange, bidentate binding of the BINAS ligand and intact Au25 clusters. The reaction products are identified as Au25(2-PET)18â2x(BINAS)x (x = 1â4) for a 24 h reaction with a 50-fold molar excess of BINAS. Two likely binding motifs are discussed. Analysis of atomic distances in both the cluster and the free ligand indicates interstaple binding connecting the central sulfur atom of the protecting (SRAu)2SR with the outer sulfur atom of a second unit. The results presented have implications on the binding position of BINAS in Au38(SR)24â2x(BINAS)x clusters.
On the flexibility of the gold-thiolate interface: Racemization of the Au40(SR)24 cluster
B. Varnholt, I. Dolamic, S. Knoppe and T. Bürgi Nanoscale, 5 (2013), p9568-9571
DOI:10.1039/c3nr03389a | unige:30154 | Abstract | Article PDF
The two enantiomers of the Au40(2-PET)24 cluster were collected using HPLC and analyzed by MALDI-TOF mass spectrometry, UV-vis- and CD-spectroscopy. The flexibility of the cluster surface allows racemization of the intrinsically chiral cluster at elevated temperatures (80 â 130 °C) which was monitored following the optical activity. The determined activation energy (25 kcal/mol) lies in the range of previously reported values for Au38 nanoclusters whereas the activation entropy deviates significantly from the one in Au38. The latter may indicate that the racemization can take place via different mechanisms.
A new multiconfigurational quantum chemical method, SplitGAS, is presented. The configuration interaction expansion, generated from a generalized active space, GAS, wave function is split in two parts, a principal part containing the most relevant configurations and an extended part containing less relevant, but not negligible, configurations. The partition is based on an orbital criterion. The SplitGAS method has been employed to study the HF, N2, and Cr2 molecules. The results on these systems, especially on the challenging, multiconfigurational Cr2 molecule, are satisfactory. While SplitGAS is comparable with the GASSCF method in terms of memory requirements, it performs better than the complete active space method followed by second-order perturbation theory, CASPT2, in terms of equilibrium bond length, dissociation energy, and vibrational properties.
Broadband ultraviolet-visible transient absorption spectroscopy in the nanosecond to microsecond time domain with sub-nanosecond time resolution
B. Lang, S. Mosquera-Vazquez, D. Lovy, P. Sherin, V. Markovic and E. Vauthey Review of Scientific Instruments, 84 (7) (2013), p73107 Keywords: delays, high-speed optical techniques, measurement by laser beam, neodymium, optical sensors, photodetectors, photoexcitation, photolysis, probes, Q-switching, solid lasers, time-domain analysis, timing jitter, ultraviolet spectroscopy, visible spectroscopy
DOI:10.1063/1.4812705 | unige:28965 | Abstract | Article HTML | Article PDF
A combination of sub-nanosecond photoexcitation and femtosecond supercontinuum probing is used to extend femtosecond transient absorption spectroscopy into the nanosecond to microsecond time domain. Employing a passively Q-switched frequency tripled Nd:YAG laser and determining the jitter of the time delay between excitation and probe pulses with a high resolution time delay counter on a single-shot basis leads to a time resolution of 350 ps in picosecond excitation mode. The time overlap of almost an order of magnitude between fs and sub-ns excitation mode permits to extend ultrafast transient absorption (TA) experiments seamlessly into time ranges traditionally covered by laser flash photolysis. The broadband detection scheme eases the identification of intermediate reaction products which may remain undetected in single-wavelength detection flash photolysis arrangements. Single-shot referencing of the supercontinuum probe with two identical spectrometer/CCD arrangements yields an excellent signal-to-noise ratio for the so far investigated chromophores in short to moderate accumulation times.
Complexes with Redox-Active Ligands: Synthesis, Structure, and Electrochemical and Photophysical Behavior of the Ru(II) Complex with TTF-Annulated Phenanthroline
L.K. Keniley, N. Dupont, L. Ray, J. Ding, K. Kovnir, J.M. Hoyt, A. Hauser and M. Shatruk Inorganic Chemistry, 52 (14) (2013), p8040-8052
DOI:10.1021/ic4006949 | unige:28963 | Abstract | Article HTML | Article PDF
Ru(II) complexes with chelating ligands, 4â²,5â²-ethylenedithiotetrathiafulvenyl[4,5-f][1,10]phenanthroline (L1), 1,3-dithiole-2-thiono[4,5-f][1,10]phenanthroline (L2), and 1,3-dithiole-2-ono[4,5-f][1,10]phenanthroline (L3), have been prepared and their structural, electrochemical, and photophysical properties investigated. Density functional theory (DFT) calculations indicate that the highest occupied molecular orbital of [Ru(bpy)2(L1)](PF6)2 (1) is located on the tetrathiafulvalene (TTF) subunit and appears â0.6 eV above the three Ru-centered d orbitals. In agreement with this finding, 1 exhibits three reversible oxidations: the two at lower potentials take place on the TTF subunit, and the one at higher potential is due to the Ru3+/Ru2+ redox couple. Complexes [Ru(bpy)2(L2)](PF6)2 (2) and [Ru(bpy)2(L3)](PF6)2 (3) exhibit only the Ru3+/Ru2+-related oxidation. The optical absorption spectra of all complexes reveal a characteristic metal-to-ligand charge transfer (MLCT) band centered around 450 nm. In addition, in the spectrum of 1 the MLCT band is augmented by a low-energy tail that extends beyond 500 nm and is attributed to the intraligand charge transfer (ILCT) transition of L1, according to time-dependent DFT calculations. The substantial decrease in the luminescence quantum yield of 1 compared to those of 2 and 3 is attributed to the reductive quenching of the emissive state via electron transfer from the TTF subunit to the Ru3+ center, thus allowing nonradiative relaxation to the ground state through the lower-lying ILCT state. In the presence of O2, complex 1 undergoes a photoinduced oxidative cleavage of the central CâC bond of the TTF fragment, resulting in complete transformation to 3. This photodegradation process was studied with 13C NMR and optical absorption spectroscopy.
Light Induced Bistability in the 2D Coordination Network {[Fe(bbtr)3](BF4)2}∞: Wavelength-Selective Addressing of Molecular Spin States
P. Chakraborty, S. Pillet, E.-E. Bendeif, C. Enachescu, R. Bronisz and A. Hauser Chemistry - A European Journal, 19 (34) (2013), p11418-11428 Keywords: cooperative effects;iron;photoinduced bistability;photoswitching;spin crossover
DOI:10.1002/chem.201301257 | unige:29524 | Abstract | Article PDF
Whereas the neat polymeric Fe(II) compound {[Fe(bbtr)3](ClO4)2}â (bbtr=1,4-di(1,2,3-triazol-1-yl)butane) shows an abrupt spin transition centered at 107 K facilitated by a crystallographic symmetry breaking, in the covalently linked 2D coordination network of {[Fe(bbtr)3](BF4)2}â, Fe(II) stays in the high-spin state down to 10 K. However, strong cooperative effects of elastic origin result in reversible, persistent and wavelength-selective photoswitching between the low-spin and high-spin manifolds. This compound thus shows true light-induced bistability below 100 K. The persistent bidirectional optical switching behavior is discussed as a function of temperature, irradiation time and intensity. Crystallographic studies reveal a photo-induced symmetry breaking and serve to establish the correlation between structure and cooperative effects. The static and kinetic behavior is explicated within the framework of the mean-field approximation.
Intrinsically chiral thiolate-protected gold clusters were recently separated into their enantiomers and their circular dichroism (CD) spectra were measured. Introduction of the chiral R-1,1â-binaphthyl-2,2â-dithiol (BINAS) into the ligand layer of rac-Au38(2-PET)24 clusters (2-PET: 2-phenylethylthiolate, SCH2CH2Ph) was shown to be diastereoselective. In this contribution, we isolated and characterized the diastereomeric reaction products of the first exchange step, A-Au38(2-PET)22(R-BINAS)1 and C-Au38(2-PET)22(R-BINAS)1 (A/C, anti-clockwise/clockwise) and the second exchange product, A-Au38(2-PET)20(R-BINAS)2. The absorption spectra show minor, but significant influence of the BINAS ligand. Overall, the spectra are less defined compared to Au38(2-PET)24, which is ascribed to symmetry breaking. The CD spectra are similar to those of the parent Au38(2-PET)24 enantiomers, readily allowing the assignment of handedness of the ligand layer. Nevertheless, some characteristic differences are found between the diastereomers. The anisotropy factors are slightly lower after ligand exchange. The second exchange step seems to confirm the trend. Inversion experiments were performed and compared to the racemization of Au38(2-PET)24. It was found that the introduction of the BINAS ligand effectively stabilizes the cluster against inversion, which involves a rearrangement of the thiolates on the cluster surface. It therefore seems that introduction of the di-thiol reduces the flexibility of the gold-sulfur interface.
Determination of the molecular structure of the short-lived light-induced high-spin state in the spin-crossover compound [Fe(6-mepy)3tren](PF6)2
P. Chakraborty, A. Tissot, L. Peterhans, L. Guénée, C. Besnard, P. Pattison and A. Hauser Physical Review B, 87 (21) (2013), p214306
DOI:10.1103/PhysRevB.87.214306 | unige:28964 | Abstract | Article PDF
In the spin-crossover compound [Fe(6-mepy)3tren](PF6)2, (6-mepy)3tren = tris{4-[(6-methyl)-2-pyridyl]-3-aza-butenyl}amine, the high-spin state can be populated as metastable state below the thermal transition temperature via irradiation into the metal to ligand charge transfer absorption band of the low-spin species. At 10 K, the lifetime of this metastable state is only 1 s. Despite this, it is possible to determine an accurate excited state structure by following the evolution of relevant structural parameters by synchrotron X-ray diffraction under continuous irradiation with increasing intensity. The difference in metal-ligand bond length between the high-spin and the low-spin state is found to be 0.192 Ã obtained from an analysis of the experimental data using the mean-field approximation to model cooperative effects.
Unambiguous evidence for the formation of excited ions upon ultrafast bimolecular photoinduced charge separation is found using a combination of femtosecond time-resolved fluorescence up-conversion, infrared and visible transient absorption spectroscopy. The reaction pathways are tracked by monitoring the vibrational energy redistribution in the product after charge separation and subsequent charge recombination. For moderately exergonic reactions, both donor and acceptor are found to be vibrationally hot, pointing to an even redistribution of the energy dissipated upon charge separation and recombination in both reaction partners. For highly exergonic reactions, the donor is very hot, whereas the acceptor is mostly cold. The asymmetric energy redistribution is due to the formation of the donor cation in an electronic excited state upon charge separation, confirming one of the hypotheses for the absence of the Marcus inverted region in photoinduced bimolecular charge separation processes
A modified cyclen azaxanthone ligand as a new fluorescent probe for Zn2+
H. Nouri, C. Cadiou, L.M. Lawson Daku, A. Hauser, S. Chevreux, I. Déchamps-Olivier, F. Lachaud, R. Ternane, M. Trabelsi-Ayadi, F. Chuburu and G. Lemercier Dalton Transactions, 42 (2013), p12157-12164
DOI:10.1039/c3dt51216a | unige:29225 | Abstract | Article PDF
A new cyclen derivative L, bearing a methyl-chromeno-pyridinylidene hydrazone moiety, was synthesized and studied in MeOH, as potential fluorescent âOFF-on-ONâ sensors for Zn(II). Photocphysical properties of this ligand being PET regulated, L was only weakly emissive in the absence of metal ions (OFF). L fluorescence was increased modestly upon addition of one equivalent of Zn(II), and further increased upon addition of a second equivalent. Therefore, Zn:L behaved as a highly sensitive ON sensor for zinc. This efficiency was correlated to Zn(II) coordination via the hydrazone moiety of the fluorophore, producing an efficient CHelation-Enhanced Fluorescence (CHEF) effect. A complementary theoretical study carried out with DFT calculations further elucidated of the optical properties.
A switch in time: A fast precipitation technique was used to prepare 75 nm FeII spin-crossover nanocrystals. Their photoswitching dynamics, based on the light-induced excited spin-state trapping effect, has been investigated by means of optical spectroscopy. A significant variation of the switching proprieties is observed compared to similar but amorphous nanoparticles.
Stimuli Responsive Hybrid Magnets: Tuning the Photoinduced Spin-Crossover in Fe(III) Complexes Inserted into Layered Magnets
M. Clemente-León, E. Coronado, M. López-Jordà, J.C. Waerenborgh, C. Desplanches, H. Wang, J.-F. Létard, A. Hauser and A. Tissot Journal of the American Chemical Society, 135 (23) (2013), p8655-8667
DOI:10.1021/ja402674x | unige:28703 | Abstract | Article HTML | Article PDF
The insertion of a [Fe(sal2-trien)]+ complex cation into a 2D oxalate network in the presence of different solvents results in a family of hybrid magnets with coexistence of magnetic ordering and photoinduced spin-crossover (LIESST effect) in compounds [FeIII(sal2-trien)][MnIICrIII(ox)3]·CHCl3 (1·CHCl3), [FeIII(sal2-trien)][MnIICrIII(ox)3]·CHBr3 (1·CHBr3), and [FeIII(sal2-trien)][MnIICrIII(ox)3]·CH2Br2 (1·CH2Br2). The three compounds crystallize in a 2D honeycomb anionic layer formed by MnII and CrIII ions linked through oxalate ligands and a layer of [Fe(sal2-trien)]+ complexes and solvent molecules (CHCl3, CHBr3, or CH2Br2) intercalated between the 2D oxalate network. The magnetic properties and Mössbauer spectroscopy indicate that they undergo long-range ferromagnetic ordering at 5.6 K and a spin crossover of the intercalated [Fe(sal2-trien)]+ complexes at different temperatures T1/2. The three compounds present a LIESST effect with a relaxation temperature TLIESST inversely proportional to T1/2. The isostructural paramagnetic compound, [FeIII(sal2-trien)][ZnIICrIII(ox)3]·CH2Cl2 (2·CH2Cl2) was also prepared. This compound presents a partial spin crossover of the inserted FeIII complex as well as a LIESST effect. Finally, spectroscopic characterization of the FeIII doped compound [Ga0.99Fe0.01(sal2trien)][MnIICrIII(ox)3]·CH2Cl2 (3·CH2Cl2) shows a gradual and complete thermal spin crossover and a LIESST effect on the isolated FeIII complexes. This result confirms that cooperativity is not a necessary condition to observe the LIESST effect in an FeIII compound.
Extracting Information about Chemical Bonding from Molecular Electron Densities via Single Exponential Decay Detector (SEDD)
P. De Silva, J. Korchowiec, N.J.S. Ram and T.A. Wesolowski Chimia, 67 (4) (2013), p253-256 Keywords: aromaticity; bonding; conceptual DFT; electron density; electron localization
DOI:10.2533/chimia.2013.253 | unige:28399 | Abstract | Article PDF
The recently introduced molecular descriptor (Single Exponential Decay Detector - SEDD) [P. de Silva, J. Korchowiec, T. A. Wesolowski, ChemPhysChem 2012, 13, 3462] is used to visualize bonding patterns in molecules. In each point of space SEDD is simply related to the electron density:
SEDD(r) = ln[1/Ï2(â(âÏ/Ï)2)2}.
Either experimental or computed densities Ï(r) can be used to evaluate SEDD. Here, maps of SEDD are obtained from theoretical densities and reveal such features as core electrons, chemical bonds, lone pairs and delocalization in aromatic systems. It is shown that SEDD provides fingerprints of aromaticity, which can be separated into geometric and electronic effects.
Non-additive kinetic energy and potential in analytically solvable systems and their approximated counterparts
T.A. Wesolowski and A. Savin
in "Recent Progress in Orbital-free Density Functional Theory (Recent Advances in Computational Chemistry Vol. 6)"
T.A. Wesolowski and Y.A. Wang Eds., World Scientific,
6 (2013), p275-295
DOI:10.1142/9789814436731_0009 | unige:28400 | Abstract | Article PDF
The one-electron equation for orbitals embedded in frozen electron density (Eqs. 20-21 in [Wesolowski and Warshel, J. Phys. Chem, 97 (1993) 8050]) in its exact and approximated version is solved for an analytically solvable model system. The system is used to discuss the role of the embedding potential in preventing the collapse of a variationally obtained electron density onto the nucleus in the case when the frozen density is chosen to be that of the innermost shell. The approximated potential obtained from the second-order gradient expansion for the kinetic energy prevents such a collapse almost perfectly but this results from partial compensation of flaws of its components. It is also shown that that the quality of a semi-local approximation to the kinetic-energy functional, a quantity needed in orbital-free methods, is not related to the quality of the non-additive kinetic energy potential - a key component of the effective embedding potential in one-electron equations for embedded orbitals.
Semilocal approximations for the kinetic energy
F. Tran and T.A. Wesolowski
in "Recent Progress in Orbital-free Density Functional Theory (Recent Advances in Computational Chemistry Vol. 6)"
T.A. Wesolowski and Y.A. Wang Eds., World Scientific,
6 (2013), p429-442
DOI:10.1142/9789814436731_0016 | unige:28401 | Abstract | Article PDF
Approximations to the non-interacting kinetic energy Ts[Ï], which take the form of semilocal analytic expressions are collected. They are grouped according to the quantities on which they explicitly depend. Additionally, the approximations for quantities related to Ts[Ï] (kinetic potential and non-additive kinetic energy), for which the analytic expressions for the âparentâ approximation for the functional Ts[Ï] are unknown, are also given.
Four-dimensional (4D) electron microscopy (EM) uniquely combines the high spatial resolution to pinpoint individual nano-objects, with the high temporal resolution necessary to address the dynamics of their laser-induced transformation. Here, using 4D-EM, we demonstrate the in situ irreversible transformation of individual nanoparticles of the molecular framework Fe(pyrazine)Pt(CN)4. The newly formed material exhibits an unusually large negative thermal expansion (i.e. contraction), which is revealed by time-resolved imaging and diffraction. Negative thermal expansion is a unique property exhibited by only few materials. Here we show that the increased flexibility of the metalâcyanide framework after the removal of the bridging pyrazine ligands is responsible for the negative thermal expansion behavior of the new material. This in situ visualization of single nanostructures during reactions should be extendable to other classes of reactive systems.
Chiral thiolate-protected gold clusters of atomic precision have gained increasing interest in recent years due to their potential use in catalysis, sensing or bioapplications. While the protection of gold clusters with chiral ligands is a rather trivial task, it was found that the clusters can bear intrinsically chiral features, most obvious in the arrangement of the protecting ligands on the surface of the cluster. Recent efforts showed the separation of the enantiomers of such intrinsically chiral gold clusters. This technique can be used for the prediction of chirality in structurally unknown clusters. Activation barriers for the racemization of Au38(SR)24 were determined. As this involves a huge rearrangement of the ligands, the flexibility of the gold-thiolate interface is demonstrated. Furthermore, the ligand exchange reactions between intrinsically chiral clusters and bidentate chiral thiols were studied. A limited, regioselective exchange was found. Most importantly, the reaction is diastereoselective and allows tailoring of gold clusters that are protected with a defined layer of ligands.
The advancement of techniques that can probe the behaviour of individual nanoscopic objects is of paramount importance in various disciplines, including photonics and electronics. As it provides images with a spatiotemporal resolution, four-dimensional electron microscopy, in principle, should enable the visualization of single-nanoparticle structural dynamics in real and reciprocal space. Here, we demonstrate the selectivity and sensitivity of the technique by visualizing the spin crossover dynamics of single, isolated metalâorganic framework nanocrystals. By introducing a small aperture in the microscope, it was possible to follow the phase transition and the associated structural dynamics within a single particle. Its behaviour was observed to be distinct from that imaged by averaging over ensembles of heterogeneous nanoparticles. The approach reported here has potential applications in other nanosystems and those that undergo (bio)chemical transformations.
The crystal structures of the M2NaIO6 series (M = Ca, Sr, Ba), prepared at 650 °C by ceramic methods, were determined from conventional laboratory X-ray powder diffraction data. Synthesis and crystal growth were made by oxidizing Iâ with O2(air) to I7+ followed by crystal growth in the presence of NaF as mineralizator, or by the reaction of the alkali-metal periodate with the alkaline-earth metal hydroxide. All three compounds are insoluble and stable in water. The barium compound crystallizes in the cubic space group Fm3m (no. 225) with lattice parameters of a = 8.3384(1) à , whereas the strontium and calcium compounds crystallize in the monoclinic space group P21/c (no. 14) with a = 5.7600(1) à , b = 5.7759(1) à , c = 9.9742(1) à , β = 125.362(1)° and a = 5.5376(1) à , b = 5.7911(1) à , c = 9.6055(1) à , β = 124.300(1)°, respectively. The crystal structure consists of either symmetric (for Ba) or distorted (for Sr and Ca) perovskite superstructures. Ba2NaIO6 contains the first perfectly octahedral [IO6]5â unit reported. The compounds of the ortho-periodates are stable up to 800 °C. Spectroscopic measurements as well as DFT calculations show a reasonable agreement between calculated and observed IR- and Raman-active vibrations.
Tetrathiafulvalene-1,3,5-triazines as (Multi)Donor-Acceptor Systems with Tunable Charge Transfer: Structural, Photophysical, and Theoretical Investigations
F. Pop, F. Riobé, S. Seifert, T. Cauchy, J. Ding, N. Dupont, A. Hauser, M. Koch and N. Avarvari Inorganic Chemistry, 52 (9) (2013), p5023-5034
DOI:10.1021/ic3027336 | unige:27865 | Abstract | Article HTML | Article PDF
Palladium-catalyzed cross-coupling reactions between chlorinated 1,3,5-triazines (TZ) and tetrathiafulvalene (TTF) trimethyltin derivatives afford mono- and C3 symmetric tris(TTF)-triazines as donorâacceptor compounds in which the intramolecular charge transfer (ICT) is modulated by the substitution scheme on TTF and TZ and by chemical or electrochemical oxidation. The TTF-TZ-Cl2 and (SMe)2TTF-TZ-Cl2 derivatives show fully planar structures in the solid state as a consequence of the conjugation between the two units. Electrochemical and photophysical investigations, supported by theoretical calculations, clearly demonstrate that the lowest excited state can be ascribed to the intramolecular charge transfer (ICT) Ï(TTF)âÏ*(TZ) transition. The tris(TTF) compound [(SMe)2TTF]3-TZ shows fluorescence when excited in the ICT band, and the emission is quenched upon oxidation. The radical cations TTF+â¢Â are easily observed in all of the cases through chemical and electrochemical oxidation by steady-state absorption experiments. In the case of [(SMe)2TTF]3-TZ, a low energy band at 5000 cmâ1, corresponding to a coupling between TTF+â¢Â and TTF units, is observed. A crystalline radical cation salt with the TTF-TZ-Cl2 donor and PF6â anion, prepared by electrocrystallization, is described.
Ultrafast photochemical processes can occur in parallel with the relaxation of the optically populated excited state toward equilibrium. The latter involves both intra- and intermolecular modes, namely vibrational and solvent coordinates, and takes place on timescales ranging from a few tens of femtoseconds to up to hundreds of picoseconds, depending on the system. As a consequence, the reaction dynamics can substantially differ from those usually measured with slower photoinduced processes occurring from equil-ibrated excited states. For example, the decay of the excited-state population may become strongly nonexponential and depend on the excitation wavelength, contrary to the Kasha and Vavilov rules. In this article, we first give a brief account of our current understanding of vibrational and solvent relaxation processes. We then present an overview of important classes of ultrafast photochemical reactions, namely electron and proton transfer as well as isomerization, and illustrate with several examples how nonequilibrium effects can affect their dynamics.
Chiral Selectivity in the Binding of [4]Helicene Derivatives to Double-Stranded DNA
O. Kel, A. Fürstenberg, N. Mehanna, C. Nicolas, B. Laleu, M. Hammarson, B. Albinsson, J. Lacour and E. Vauthey Chemistry - A European Journal, 19 (22) (2013), p7173-7180 Keywords: chirality;circular dichroism;DNA recognition;fluorescent probes;helical structures;time-resolved spectroscopy
DOI:10.1002/chem.201203915 | unige:27926 | Abstract | Article HTML | Article PDF
The interaction of a series of chiral cationic [4]helicene derivatives, which differ by their substituents, with double-stranded DNA has been investigated by using a combination of spectroscopic techniques, including time-resolved fluorescence, fluorescence anisotropy, and linear dichroism. Addition of DNA to helicene solutions results to a hypochromic shift of the visible absorption bands, an increase of fluorescence quantum yield and lifetime, a slowing down of fluorescence anisotropy decay, and a linear dichroism in flow-oriented DNA, which unambiguously points to the binding of these dyes to DNA. Both helicene monomers and dimeric aggregates, which form at higher concentration, bind to DNA, the former most probably upon intercalation and the latter upon groove binding. The binding constant depends substantially on the dye substituents and is, in all cases, larger with the M than the P enantiomer, by factors ranging from 1.2 to 2.3, depending on the dye.
The time resolution of photon detection systems is important for a wide range of applications in physics and chemistry. It impacts the quality of time-resolved spectroscopy of ultrafast processes and has a direct influence on the best achievable time resolution of time-of-flight detectors in high-energy and medical physics. For the characterization of photon detectors, it is important to measure their exact timing properties in dependence of the photon flux and the operational parameters of the photodetector and its accompanying electronics. We report on the timing of silicon photomultipliers (SiPM) as a function of their bias voltage, electronics threshold settings and the number of impinging photons. We used ultrashort laser pulses at 400 nm wavelength with pulse duration below 200 fs. We focus our studies on different types of SiPMs (Hamamatsu MPPC S10931-025P, S10931-050P and S10931-100P) with different SPAD sizes (25μm, 50μm and 100μm) coupled to the ultrafast discriminator amplifier NINO. For the SiPMs, an optimum in the time resolution regarding bias and threshold settings can be reached. For the 50μm type, we achieve a single photon time resolution of 80 ps sigma, and for saturating photon fluxes better than 10 ps sigma.
The photophysics and photochemistry of kynurenic acid (KNA) and kynurenine yellow (KNY) in neutral aqueous solutions were investigated using time-resolved optical spectroscopy. Both molecules have similar quinoline-like structures, the only difference being the absence of conjugation in the nitrogen containing cycle in KNY. The main channel of S1 excited state decay in the case of partially-unconjugated KNY is the solvent assisted S1 â S0 radiationless transition via intermolecular hydrogen bonds (ΦIC = 0.96), whereas, in the case of fully-conjugated KNA, it is intersystem crossing to the triplet state (ΦT = 0.82). The major intermediate products of the singlet excited KNY deactivation are the triplet state (ΦT = 0.022) and, most probably, the enol form (Φenol = 0.012), which decay with the formation of 2,3-dihydro-4-hydroxyquinoline and 4-hydroxyquinoline, respectively. The results obtained show that KNA and KNY, which are products of the decomposition of the UV filter kynurenine, are significantly more photoactive and less photostable than the parent molecule.
Effect of Temperature and Pressure on Emission Lifetime of Sm2+ Ion Doped in MFX (M=Sr, Ba; X=Br, I) Crystals
P. Pal, T. Penhouët, V. D'Anna and H. Hagemann Journal of Luminescence, 142 (2013), p66-74 Keywords: Sm2+; high pressure lifetime; temperature lifetime; SrFBr; BaFBr; BaFI
DOI:10.1016/j.jlumin.2013.03.011 | unige:27714 | Abstract | Article PDF
The emission lifetime of Sm2+ ions doped in MFX (M=Ba, Sr; X=Br, I) crystals was investigated as a function of pressure and temperature. The decay of the 5DJ(J=0,1,2) levels showed single exponential relaxation. The analysis of these experiments yielded the position of the lowest 4f55d1 state as well as non-radiative rate constants. These values were compared with those for Sm2+ doped in other matlockite host crystals. The single exponential decrease of the 5D0,1 lifetime as a function of pressure was described considering the increased radiative decay rates of these 5D0,1 levels through electronic mixing between the 4f55d1 and 5DJstates.
Study of Surfactant Alcohols with various Chemical Motives at the Hydrophilic/Hydrophobic Interface
P.-L. Zaffalon, V. D'Anna, H. Hagemann and A. Zumbuehl RSC Advances, 3 (20) (2013), p7237-7244
DOI:10.1039/c3ra40704g | unige:27864 | Abstract | Article PDF
The melting behavior, the solubility, and the influence of hydrogen bonds were analyzed for a series of single- and double-tailed surfactant alcohols. Various effects such as the presence of free amides or the intermolecular spacing were found to be important factors for increasing or decreasing the melting temperature of a surfactant. Furthermore, we present a model for the packing of diamido-lipids and study the temperature-dependence of the IR signals.
A comparison of the vibrational spectra of many inorganic borohydrides allows us to distinguish compounds with isolated BH4- ions and compounds containing complex ions such as Sc(BH4)4-. The characteristic spectral features of both types of compounds are identified, showing that the BâH bonding is quite different in both cases. A detailed analysis of the vibrations of the isolated BH4-Â ions provides new information about their local structure. Angular deformations of individual borohydride ion are analyzed quantitatively. It appears that the compounds containing isolated BH4-Â ions belong to those with the most electropositive cations and the highest decomposition temperature, while the complex borohydrides show significantly lower decomposition temperatures and possible diborane formation.
Synthesis and Redox and Photophysical Properties of Benzodifuran–Spiropyran Ensembles
H. Li, J. Ding, S. Chen, C. Beyer, S.-X. Liu, H.-A. Wagenknecht, A. Hauser and S. Decurtins Chemistry - A European Journal, 19 (20) (2013), p6459-6466 Keywords: asymmetric synthesis;click chemistry;electron transfer;photochromism;redox chemistry;spiro compounds
DOI:10.1002/chem.201204043 | unige:27859 | Abstract | Article PDF
Two benzodifuran (BDF)-coupled spiropyran (SP) systems and their BDF reference compounds were obtained in good yields through HuisgenâMeldalâSharpless âclickâ chemistry and then subjected to investigation of their electrochemical and photophysical properties. In both SP and merocyanine (MC) forms of the coupled molecules, the BDF-based emission is quenched to around 1â% of the quantum yield of emission from the BDF reference compounds. Based on electrochemical data, this quenching is attributed to oxidative electron-transfer quenching. Irradiation at 366â nm results in ring opening to the MC forms of the BDF-coupled SP compounds and the SP reference compound with a quantum efficiency of about 50â%. The rate constants for the thermal ring closing are approximately 3.4Ã10â3 sâ1. However, in the photostationary states the MC fractions of the coupled molecules are substantially lower than that of the reference SP compound, attributed to the observed acceleration of the ring-closing reaction upon irradiation. As irradiation at 366â nm invariably also excites higher-energy transitions of the BDF units in the coupled compounds, the ring-opening reaction is accelerated relative to the SP reference, which results in lower MC fractions in the photostationary state. Reversible photochromism of these BDF-coupled SP compounds renders them promising in the field of molecular switches.
The structurally characterized tetrathiafulvalene-1,2,4,5-tetrazine donorâacceptor system shows redox tuneable intramolecular charge transfer, solvatochromic and electrochromic behaviour. Attachment of a dipicolyl-amine chelating unit affords a multifunctional ligand, which allows the preparation of the ZnCl2 complex in which an anion-Ï interaction is seen.
Electron Transfer between Hydrogen-Bonded Pyridylphenols and a Photoexcited Rhenium(I) Complex
W. Herzog, C. Bronner, S. Löffler, B. He, D. Kratzert, D. Stalke, A. Hauser and O.S. Wenger ChemPhysChem, 14 (6) (2013), p1168-1176 Keywords: electron transfer;luminescence;photochemistry;proton transfer;transient absorption
DOI:10.1002/cphc.201201069 | unige:27484 | Abstract | Article PDF
Two pyridylphenols with intramolecular hydrogen bonds between the phenol and pyridine units have been synthesized, characterized crystallographically, and investigated by cyclic voltammetry and UV/Vis spectroscopy. Reductive quenching of the triplet metal-to-ligand charge-transfer excited state of the [Re(CO)3(phen)(py)]+Â complex (phen=1,10-phenanthroline, py=pyridine) by the two pyridylphenols and two reference phenol molecules is investigated by steady-state and time-resolved luminescence spectroscopy, as well as by transient absorption spectroscopy. SternâVolmer analysis of the luminescence quenching data provides rate constants for the bimolecular excited-state quenching reactions. H/D kinetic isotope effects for the pyridylphenols are on the order of 2.0, and the bimolecular quenching reactions are up to 100â times faster with the pyridylphenols than with the reference phenols. This observation is attributed to the markedly less positive oxidation potentials of the pyridylphenols with respect to the reference phenols (â0.5â V), which in turn is caused by proton coupling of the phenol oxidation process. Transient absorption spectroscopy provides unambiguous evidence for the photogeneration of phenoxyl radicals, that is, the overall photoreaction is clearly a proton-coupled electron-transfer process.
We report a detailed DFT study of the energetic and structural properties of the spin-crossover Co(II) complex [Co(tpy)2]2+ (tpy = 2,2â²:6â²,2â²â²-terpyridine) in the low-spin (LS) and the high-spin (HS) states, using several generalized gradient approximation and hybrid functionals. In either spin-state, the results obtained with the functionals are consistent with one another and in good agreement with available experimental data. Although the different functionals correctly predict the LS state as the electronic ground state of [Co(tpy)2]2+, they give estimates of the HSâLS zero-point energy difference ÎE0HL (tpy)  which strongly depend on the functional used. This dependency on the functional was also reported for the DFT estimates of the zero-point energy difference ÎE0HL (bpy)  in the HS complex [Co(bpy)3]2+ (bpy = 2,2â²-bipyridine) [A. Vargas, A. Hauser and L. M. Lawson Daku, J. Chem. Theory Comput., 2009, 5, 97]. The comparison of the ÎE0HL (tpy)  and ÎE0HL (bpy)  estimates showed that all functionals correctly predict an increase of the zero-point energy difference upon the bpy â tpy ligand substitution, which furthermore weakly depends on the functionals, amounting to (ÎE0HL)bpy->tpy â +2670 cm-1 . From these results and basic thermodynamic considerations, we establish that, despite their limitations, current DFT methods can be applied to the accurate determination of the spin-state energetics of complexes of a transition metal ion, or of these complexes in different environments, provided that the spin-state energetics is accurately known in one case. Thus, making use of the availability of a highly accurate ab initio estimate of the HSâLS energy difference in the complex [Co(NCH)6]2+ [L. M. Lawson Daku, F. Aquilante, T. W. Robinson and A. Hauser, J. Chem. Theory Comput., 2012, 8, 4216], we obtain for [Co(tpy)2]2+ and [Co(bpy)3]2+best estimates of ÎE0HL (bpy) â -2800 cm-1  and ÎE0HL (tpy) â 0 cm-1 , in good agreement with the known magnetic behaviour of the two complexes.
A Straightforward Synthesis and Structure-Activity Relationship of Highly Efficient Initiators for Two-Photon Polymerization
Z. Li, N. Pucher, K. Cicha, J. Torgersen, S.C. Ligon, A. Ajami, W. Husinsky, A. Rosspeintner, E. Vauthey, S. Naumov, T. Scherzer, J. Stampfl and R. Liska Macromolecules, 46 (2) (2013), p352-361
DOI:10.1021/ma301770a | unige:26500 | Abstract | Article HTML | Article PDF | Video
The development of practical two-photon absorption photoinitiators (TPA PIs) has been slow due to their complicated syntheses often reliant on expensive catalysts. These shortcomings have been a critical obstruction for further advances in the promising field of two-photon-induced photopolymerization (TPIP) technology. This paper describes a series of linear and cyclic benzylidene ketone-based two-photon initiators containing double bonds and dialkylamino groups synthesized in one step via classical aldol condensation reactions. Systematic investigations of structureâactivity relationships were conducted via quantum-chemical calculations and experimental tests. These results showed that the size of the central ring significantly affected the excited state energetics and emission quantum yields as well as the two-photon initiation efficiency. In the TPIP tests the 4-methylcyclohexanone-based initiator displayed much broader ideal processing windows than its counterparts with a central five-membered ring and previously described highly active TPA PIs. Surprisingly, a writing speed as high as 80 mm/s was obtained for the microfabrication of complex 3D structures employing acrylate-based formulations. These highly active TPA PIs also exhibit excellent thermal stability and remain inert to one-photon excitation. Straightforward synthesis combined with high TPA initiation efficiency makes these novel initiators promising candidates for commercialization.
The excited-state dynamics of two energy donorâbridgeâacceptor (DâBâA) systems consisting of a zinc tetraphenylporphyrin (ZnP) and a free base tetraphenylporphyrin (FbP) bridged by oligo-p-phenyleneethynylene units with different substituents has been investigated using ultrafast spectroscopy. These systems differ by the location of the lowest singlet excited state of the bridge, just above or below the S2Â porphyrin states. In the first case, Soret band excitation of the porphyrins is followed by internal conversion to the local S1Â state of both molecules and by a S1Â energy transfer from the ZnP to the FbP end on the 10 ns time scale, as expected for a center-to-center distance of about 4.7 nm. On the other hand, if the bridge is excited, the energy is efficiently transferred within 1 ps to both porphyrin ends. Selective bridge excitation is not possible with the second system, because of the overlap of the absorption bands. However, the time-resolved spectroscopic data suggest a reversible conversion between the D*(S2)âBâA and DâB*(S1)âA states as well as a transition from the DâB*(S1)âA to the DâBâA* states on the picosecond time scale. This implies that the local S2energy of the ZnP end can be transported stepwise to the FbP end, i.e., over about 4.7 nm, within 1 ps with an efficiency of more than 0.2.
Improved photoluminescence and afterglow of CaTiO3:Pr3+ by ammonia treatment
S. Yoon, E.H. Otal, A.E. Maegli, L. Karvonen, S.K. Matam, S. Riegg, S.G. Ebbinghaus, J.C. Fallas, H. Hagemann, B. Walfort, S. Pokrant and A. Weidenkaff Optical Materials Express, 3 (2) (2013), p248-259 Keywords: fluorescent materials; rare-earth doped materials
DOI:10.1364/OME.3.000248 | unige:26402 | Abstract | Article HTML | Article PDF
The phosphor CaTiO3:Pr3+ was synthesized via a solid-state reaction in combination with a subsequent annealing under flowing NH3. Comparatively large off-center displacements of Ti in the TiO6 octahedra were confirmed for as-synthesized CaTiO3:Pr3 by XANES. Raman spectroscopy showed that the local crystal structure becomes highly symmetric when the powders are ammonolyzed at 400 °C. Rietveld refinement of powder X-ray diffraction data revealed that the samples ammonolyzed at 400 °C have the smallest lattice strain and at the same time the largest average Ti-O-Ti angles were obtained. The samples ammonolyzed at 400 °C also showed the smallest mass loss during the thermal re-oxidation in thermogravimetric analysis (TGA). Enhanced photolumincescence brightness and an improved decay curve as well as the highest reflectance were obtained for the samples ammonolyzed at 400 °C. The improved photoluminescence and afterglow by NH3 treatment are explained as a result of the reduced concentration of oxygen excesses with simultaneous relaxation of the lattice strain.
Tetrathiafulvalene-Benzothiadiazoles as Redox-Tunable Donor-Acceptor Systems: Synthesis and Photophysical Study
F. Pop, A. Amacher, N. Avarvari, J. Ding, L.M. Lawson Daku, A. Hauser, M. Koch, J. Hauser, S.-X. Liu and S. Decurtins Chemistry - A European Journal, 19 (7) (2013), p2504-2514 Keywords: charge transfer;donor-acceptor systems;fluorescence;photophysics;redox chemistry
DOI:10.1002/chem.201202742 | unige:26401 | Abstract | Article PDF
Electrochemical and photophysical analysis of new donorâacceptor systems 2 and 3, in which a benzothiadiazole (BTD) unit is covalently linked to a tetrathiafulvalene (TTF) core, have verified that the lowest excited state can be ascribed to an intramolecular-charge-transfer (ICT) Ï(TTF)âÏ*(benzothiadiazole) transition. Owing to better overlap of the HOMO and LUMO in the fused scaffold of compound 3, the intensity of the 1ICT band is substantially higher compared to that in compound 2. The corresponding CT fluorescence is also observed in both cases. The radical cation TTF+. is easily observed through chemical and electrochemical oxidation by performing steady-state absorption experiments. Interestingly, compound 2 is photo-oxidized under aerobic conditions.
Modular Synthesis, Orthogonal Post-Functionalization, Absorption, and Chiroptical Properties of Cationic [6]Helicenes
F. Torricelli, J. Bosson, C. Besnard, M. Chekini, T. Bürgi and J. Lacour Angewandte Chemie International Edition, 52 (6) (2013), p1796-1800 Keywords: arenes;helical structures;synthetic methods;UV/Vis spectroscopy;vicarious nucleophilic substitution
DOI:10.1002/anie.201208926 | unige:26214 | Abstract | Article HTML | Article PDF
Novel cationic diaza-, azaoxo-, and dioxo[6]helicenes are readily prepared and functionalized selectively by orthogonal aromatic electrophilic and vicarious nucleophilic substitutions (see scheme). Reductions, cross-coupling, or condensation reactions introduce additional diversity and allow tuning of the absorption properties up to the near-infrared region. The diaza salts can be resolved into single enantiomers.
Analysis of the Experimental Data for Pure and Diluted [FexZn1–x(bbtr)3](ClO4)2 Spin-Crossover Solids in the Framework of a Mechanoelastic Model
P. Chakraborty, C. Enachescu and A. Hauser European Journal of Inorganic Chemistry, 2013 (5-6) (2013), p770-780 Keywords: spin crossover;mechanoelastic model;doping;iron;zinc
DOI:10.1002/ejic.201201193 | unige:26499 | Abstract | Article PDF
The mechanoelastic model is applied to reproduce the experimental relaxation and thermal transition curves as determined for crystals of pure and diluted {[FexZn1âx(bbtr)3](ClO4)2}â [bbtr = 1,4-di(1,2,3-triazol-1-yl)butane] spin-crossover systems. In the mechanoelastic model, the spin-crossover complexes are situated in a hexagonal planar lattice, which is similar to the 2D coordination polymer with (3,6) network topology of [Fe(bbtr)3](ClO4)2. These complexes are linked by springs, which simulate the elastic interactions between them. Owing to the change in volume of the complexes during the spin transition, an elastic force accompanies the switch of every complex. This force propagates through the entire lattice and causes a shift of all molecules in the system and thus results in a new nuclear configuration. First, the ability of the model to reproduce various shapes of thermal transition and relaxation curves in pure compounds is analyzed; these range from gradual to very steep and include hysteresis behavior for the former and from single exponential to sigmoidal or with several steps for the latter. A structural phase transition can also be accounted for by changing the shape of the sample at a fixed temperature from a regular to an elongated hexagon. Furthermore, the effect of adding Zn as a dopant in a mixed crystal series is discussed. The role of dopants on the cluster evolution is also analyzed directly and by using the correlation factor.
A Donor–Acceptor Tetrathiafulvalene Ligand Complexed to Iron(II): Synthesis, Electrochemistry, and Spectroscopy of [Fe(phen)2(TTF-dppz)](PF6)2
N. Dupont, Y.-F. Ran, S.-X. Liu, J. Grilj, E. Vauthey, S. Decurtins and A. Hauser Inorganic Chemistry, 52 (1) (2013), p306-312
DOI:10.1021/ic3019277 | unige:25113 | Abstract | Article HTML | Article PDF
The synthesis and photophysical properties of the complex [Fe(phen)2(TTF-dppz)]2+Â (TTF-dppz = 4â²,5â²-bis-(propylthio)tetrathiafulvenyl[i]dipyrido[3,2-a:2â²,3â²-c]phenazine, phen = 1,10-phenanthroline) are described. In this complex, excitation into the metalâligand charge transfer bands results in the population of a high-spin state of iron(II), with a decay lifetime of approximately 1.5 ns, in dichloromethane, at room temperature. An intraligand charge transfer state can also be obtained and has a lifetime of 38 ps. A mechanism for the different states reached is proposed based on transient absorption spectroscopy.
The excited-state dynamics of two triads consisting of a naphthalenediimide (cNDI) substituted at the core by two zinc (ZnP) or free-base tetraphenylporphyrins (FbP) was investigated by ultrafast fluorescence and transient absorption spectroscopy. The electronic absorption spectra of the triads are almost the composites of those of the constituents, pointing to a weak electronic coupling and to a localization of the excitation energy on one of the porphyrins. In cyclohexane, the excited-state dynamics of the triads are essentially the same as those of the individual porphyrins, with the exception of the Soret emission of the ZnP triad, whose lifetime exhibits a more than 10 fold shortening compared to ZnP. A similarly ultrafast fluorescence decay was measured in tetrahydrofuran and benzonitrile. In these two solvents, charge separation from the excited porphyrin to the cNDI was found to take place with ~1 ps and ~25 ps time constants in the ZnP and FbP triads, respectively. The build up of the charge-separated state population in the ZnP triad is independent on the excitation wavelength, indicating that charge separation takes place from the lowest singlet excited state. Charge recombination occurs with a time constant around 8 ps in both triads, i.e. is slower than charge separation in the ZnP triad but faster in the FbP triad. These differences are rationalized in terms of the driving forces for charge separation and recombination.
The synthesis, X-ray structures and photophysical properties of several new Ln(III) complexes with pyrazine-2,6-dicarboxylic acid (H2PYZ) that demonstrate excellent stability and solubility in non-aqueous solution are reported, and compared to structurally analogous complexes with pyridine-2,6-dicarboxylic acid (H2DPA). The Eu(III) and Yb(III) complexes demonstrate efficient metal centered luminescence in the visible and Near Infra-Red (NIR) regions respectively. Low temperature (77 K) phosphorescence measurements using the corresponding Gd(III) complex allowed the photophysical properties of the sensitizer to be rationalized, together with corresponding TD-DFT studies for a model complex. Lastly, we have evaluated the sensitization efficiencies for these complexes, and have undertaken femtosecond transient absorption (TA) measurements in order to evaluate the relative importance of the intersystem crossing and energy transfer processes involved with sensitized Ln(III) emission via the antennae effect.
The absorption spectrum of fluorenone in zeolite L is calculated from first-principles simulations. The broadening of each band is obtained from the explicit treatment of the interactions between the chromophore and its environment in the statistical ensemble. The comparison between the simulated and measured spectra reveals the main factors affecting the spectrum of the chromophore in hydrated zeolite L. Whereas each distinguishable band is found to originate from a single electronic transition, the bandwidth is determined by the statistical nature of the environment of the fluorenone molecule. The K+...O=C motif is retained in all conformations. Although the interactions between K+ and the fluorenone carbonyl group result in an average lengthening of the C=O bond and in a redshift of the lowest energy absorption band compared to gas phase or non-polar solvents, the magnitude of this shift is noticeably smaller than the total shift. An important factor affecting the shape of the band is fluorenoneâs orientation, which is strongly affected by the presence of water. The effect of direct interactions between fluorenone and water is, however, negligible.
The emission spectra of Sm2+ doped in BaFBr and SrFBr hosts were measured at 10 K from ambient pressure to 8 GPa. The crystal field energy levels determined from the emission spectra were used to extract the free ion parameters (Fk and ζ ) and crystal field parameters (Bqk). The variation of Fk and ζ as a function of pressure was studied systematically and was discussed in relation to the central field and symmetry restricted covalency models. The change of the spin orbit coupling parameter (ζ) with pressure for SrFBr:Sm2+ showed very different behavior than in other matlockite hosts. Moreover the variation of Bqk under pressure was studied. The pressure dependence of the Bqk was described quantitatively using the Superposition Model (SM) with the help of structural parameters as a function of pressure, obtained from periodic DFT calculations. The validity of the SM was tested for Sm2+ in BaFBr and SrFBr. It is shown that this model does not apply to SrFBr, in contrast to other matlockite host materials.
Chromium(III)-trisoxalate, a versatile building block for luminescent materials
M. Milos and A. Hauser Journal of Luminescence, 133 (2013), p15-20 Keywords: chromium(III)-tris-oxalate; energy migration; fluorescence line narrowing; spectral diffusion; persistent spectral hole burning
DOI:10.1016/j.jlumin.2011.12.053 | unige:24060 | Article HTML | Article PDF
Chromium(III)-trisoxalate,[Cr(ox)3]3- (ox = C2O42-), incorporated into polymeric networks of composition [NaCr(ox)3][MII(bpy)3] and [NaCr(ox)3][MIII(bpy)3]ClO4 (bpy= 2,2'-bipyridine, MII = Zn, Fe, Ru; MIII = Rh, Cr), results in interesting features ranging from phonon-assisted and resonant energy migration within the R1 line the 2E state to persistent spectral side-hole burning via the latter, and manifestations of specific nearest-neighbour ÏâÏ interactions between bipyridine and oxalate.
The ligand exchange reaction between racemic Au38(2-PET)24 (2-PET: 2-phenylethylthiolate) clusters and enantiopure 1,1â-binapththyl-2,2â-dithiol (BINAS) was monitored in situ using a chiral HPLC approach. In the first exchange step, a clear preference of R-BINAS towards the left-handed enantiomer of Au38(2-PET)24 is observed (about four times faster than reaction with the right-handed enantiomer). The second exchange step is drastically slowed compared to the first step. BINAS substitution deactivates the cluster for further exchange, which is attributed to (stereo)electronic effects. The results constitute the first example of a ligand exchange reaction in a thiolate-protected gold cluster with directed enrichment of a defined species in the product mixture. This may open new possibilities for the design of nanomaterials with tailored properties.
In situ ATR-IR spectroscopy study of adsorbed protein: Visible light denaturation of bovine serum albumin on TiO2
A. Bouhekka and T. Bürgi Applied Surface Science, 261 (2012), p369-374 Keywords: in situ spectroscopy; TiO2; visible light; BSA adsorption; denaturation; protein structure
DOI:10.1016/j.apsusc.2012.08.017 | unige:24517 | Abstract | Article PDF
In this work in situ Fourier transform infrared-attenuated total reflection (FTIR-ATR) spectroscopy in a flow-through cell was used to study the effect of visible light irradiation on bovine serum albumin (BSA) adsorbed on porous TiO2 films. The experiments were performed in water at concentrations of 10â6 mol/l at room temperature. The curve fitting method of the second derivative spectra allowed us to explore details of the secondary structure of pure BSA in water and conformation changes upon adsorption as well as during and after illumination by visible light. The results clearly show that visible light influences the conformation of adsorbed BSA. The appearance of a shift of the amide I band, in the original spectra, from 1653 cmâ1 to 1648 cmâ1, is interpreted by the creation of random coil in the secondary structure of adsorbed BSA. The second derivative analysis of infrared spectra permits direct quantitative analysis of the secondary structural components of BSA, which show that the percentage of α-helix decreases during visible light illumination whereas the percentage of random coil increases.
We predict and analyze density-functional theory (DFT) -based structures for the recently isolated Au40(SR)24 cluster. Combining structural information extracted from ligand-exchange reactions, circular dichroism and transmission electron microscopy leads us to propose two families of low-energy structures that have a chiral Au-S framework on the surface. These families have a common geometrical motif where a non-chiral Au26 bi-icosahedral cluster core is protected by 6 RS-Au-SR and 4 RS-Au-SR-Au-SR oligomeric units, analogously to the âDivide and Protectâ motif of known clusters Au25(SR)18-/0, Au38(SR)24 and Au102(SR)44. The strongly prolate shape of the proposed Au26 core is supported by transmission electron microscopy. Density-of-state-analysis shows that the electronic structure of Au40(SR)24 can be interpreted in terms of a dimer of two 8-electron superatoms, where the 8 shell electrons are localized at the two icosahedral halves of the metal core. The calculated optical and chiroptical characteristics of the optimal chiral structure are in a fair agreement with the reported data for Au40(SR)24.
Heteroleptic FeII Complexes of 2,2′-Biimidazole and Its Alkylated Derivatives: Spin-Crossover and Photomagnetic Behavior
H.V. Phan, P. Chakraborty, M. Chen, Y.M. Calm, K. Kovnir, L.K. Keniley, J.M. Hoyt, E.S. Knowles, C. Besnard, M.W. Meisel, A. Hauser, C. Achim and M. Shatruk Chemistry - A European Journal, 18 (49) (2012), p15805-15815 Keywords: iron complexes;LIESST effect;N ligands;photomagnetism;spin crossover
DOI:10.1002/chem.201202045 | unige:24449 | Abstract | Article PDF
Three iron(II) complexes, [Fe(TPMA)(BIM)](ClO4)2â 0.5H2O (1), [Fe(TPMA)(XBIM)](ClO4)2 (2), and [Fe(TPMA)(XBBIM)](ClO4)2 â 0.75CH3OH (3), were prepared by reactions of FeII perchlorate and the corresponding ligands (TPMA=tris(2-pyridylmethyl)amine, BIM=2,2â²-biimidazole, XBIM=1,1â²-(α,αâ²-o-xylyl)-2,2â²-biimidazole, XBBIM=1,1â²-(α,αâ²-o-xylyl)-2,2â²-bibenzimidazole). The compounds were investigated by a combination of X-ray crystallography, magnetic and photomagnetic measurements, and Mössbauer and optical absorption spectroscopy. Complex 1 exhibits a gradual spin crossover (SCO) with T1/2=190â K, whereas 2 exhibits an abrupt SCO with approximately 7â K thermal hysteresis (T1/2=196â K on cooling and 203â K on heating). Complex 3 is in the high-spin state in the 2â300â K range. The difference in the magnetic behavior was traced to differences between the inter- and intramolecular interactions in 1 and 2. The crystal packing of 2features a hierarchy of intermolecular interactions that result in increased cooperativity and abruptness of the spin transition. In 3, steric repulsion between H atoms of one of the pyridyl substituents of TPMA and one of the benzene rings of XBBIM results in a strong distortion of the FeII coordination environment, which stabilizes the high-spin state of the complex. Both 1 and 2 exhibit a photoinduced low-spin to high-spin transition (LIESST effect) at 5â K. The difference in the character of intermolecular interactions of 1 and 2 also manifests in the kinetics of the decay of the photoinduced high-spin state. For 1, the decay rate constant follows the single-exponential law, whereas for 2 it is a stretched exponential, reflecting the hierarchical nature of intermolecular contacts. The structural parameters of the photoinduced high-spin state at 50â K are similar to those determined for the high-spin state at 295â K. This study shows that N-alkylation of BIM has a negligible effect on the ligand field strength. Therefore, the combination of TPMA and BIM offers a promising ligand platform for the design of functionalized SCO complexes.
A two-fold active control of the plasmonic resonance of randomly distributed gold nanoparticles (GNPs) has been achieved. GNPs have been immobilized on an Indium Tin Oxide (ITO) coated glass substrate and then covered with a liquid crystalline compound. The system has been investigated by means of atomic force and scanning electron microscopy, revealing the presence of isolated and well distributed GNPs. The application of an external electric field to the sample has a two-fold consequence: the re-orientation of the hybrid-aligned liquid crystal layer and the formation of a carrier accumulation layer in the proximity of the ITO substrate. The refractive indices of both liquid crystal and accumulation layers are influenced by the applied field in a competitive way and produce a âdancing behaviorâ of the GNPâs plasmonic resonance spectral position.
Highly accurate estimates of the high-spin/low-spin energy difference ÎEHLel in the high-spin complexes [Fe(NCH)6]2+ and [Co(NCH)6]2+ have been obtained from the results of CCSD(T) calculations extrapolated to the complete basis set limit. These estimates are shown to be strongly influenced by scalar relativistic effects. They have been used to assess the performances of the CASPT2 method and of 30 density functionals of the GGA, meta-GGA, global hybrid, RSH and double-hybrid types. For the CASPT2 method, the results of the assessment support the proposal [Kepenekian, M.; Robert, V.; Le Guennic, B. J. Chem. Phys.2009, 131, 114702] that the ionization potentialâelectron affinity (IPEA) shift defining the zeroth-order Hamiltonian be raised from its standard value of 0.25 au to 0.50â0.70 au for the determination of ÎEHLel in Fe(II) complexes with a [FeN6] core. At the DFT level, some of the assessed functionals proved to perform within chemical accuracy (±350 cm-1) for the spin-state energetics of [Fe(NCH)6]2+, others for that of [Co(NCH)6]2+, but none of them simultaneously for both complexes. As demonstrated through a reparametrization of the CAM-PBE0 range-separated hybrid, which led to a functional that performs within chemical accuracy for the spin-state energetics of both complexes, performing density functionals of broad applicability may be devised by including in their training sets highly accurate data like those reported here for [Fe(NCH)6]2+ and [Co(NCH)6]2+.
The strong coupling between planar arrays of gold and silver nanoparticles mediated by a near-field interaction is investigated both theoretically and experimentally to provide an in-depth study of symmetry breaking in complex nanoparticle structures. The asymmetric composition allows to probe for bright and dark eigenmodes, in accordance with plasmon hybridization theory. The strong coupling could only be observed by separating the layers by a nanometric distance with monolayers of suitably chosen polymers. The bottom-up assembly of the nanoparticles as well as the stratified structures themselves gives rise to an extremely flexible system that, moreover, allows the facile variation of a number of important material parameters as well as the preparation of samples on large scales. This flexibility was used to modify the coupling distance between arrays, showing that both the positions and relative intensities of the resonances observed can be tuned with a high degree of precision. Our work renders research in the field of âplasmonic moleculesâ mature to the extent that it could be incorporated into functional optical devices.
Tautomerization in 2,7,12,17-Tetraphenylporphycene and 9-Amino-2,7,12,17-tetraphenylporphycene: Influence of Asymmetry on the Direction of the Transition Moment
P. Fita, M. Pszona, G. Orzanowska, D. Sánchez-García, S. Nonell, E. Vauthey and J. Waluk Chemistry - A European Journal, 18 (41) (2012), p13160-13167 Keywords: hydrogen transfer;porphycenes;tautomerism;transition moments
DOI:10.1002/chem.201201432 | unige:23089 | Abstract | Article PDF
Femtosecond transient absorption anisotropy studies have been performed for two porphycenes of different symmetry. In 2,7,12,17-tetraphenylporphycene, the chemical identity of two trans forms implies a change in the S0âS1 transition-moment direction upon tautomerization. Exploiting this phenomenon, the rates of double hydrogen transfer in both the S0 and S1 states (1.4Ã1012 sâ1 and 2.7Ã1011 sâ1, respectively) have been determined by performing time-resolved anisotropy studies. In the asymmetric 9-amino-2,7,12,17-tetraphenylporphycene, tautomerization occurs with a similar rate in the ground state. In the S1 state, the reaction is hindered in its vibrationally relaxed form, but the excitation spectra suggest that it may occur for an unrelaxed molecule. Unlike all porphycenes that have been studied so far, 9-amino-2,7,12,17-tetraphenylporphycene does not reveal significant changes in anisotropy owing to intramolecular double hydrogen transfer; rather, the transition-moment directions are similar in the two tautomeric forms. Analysis of the molecular orbitals allows for an explanation of the âlockingâ of the transition moments: it is due to a large splitting of the two HOMO orbitals, which retain the order of their energies in the two tautomers.
This study addresses the free energy dependence of charge recombination following photoinduced bimolecular electron transfer in three different solvents of either inert (acetonitrile and benzyl acetate) or reactive (N,N-dimethylaniline) character. Femtosecond time-resolved fluorescence and transient absorption have been used to determine the time scales for charge recombination. In pure N,N-dimethylaniline, charge recombination is found to be substantially slower than charge separation in a range of driving forces covering 1.5 eV. In all three solvents, the so-called Marcus inverted region is clearly observed for charge recombination. Additionally, the charge recombination step is found to be influenced by the solvent relaxation dynamics. A diffusion-reaction equation approach using an electron transfer model accounting for solvent relaxation is used to rationalize the experimental results.
A Spectroscopic and Computational Study of a Photoinduced Cross-Dehydrogenative Coupling Reaction of a Stable Semiquinone Radical
J. Grilj, T.K. Todorova, C. Yi, S.-X. Liu, E. Vauthey and S. Decurtins Chemistry - A European Journal, 18 (43) (2012), p13605-13608 Keywords: density functional calculations;photochemistry;radicals;transient absorption;transition states
DOI:10.1002/chem.201201463 | unige:23743 | Abstract | Article PDF
Norrish-type-II reaction on a semiquinone radical: Stable semiquinone radicals serve as novel molecular platforms on which a Norrish-type-II photoreaction can be initiated. A detailed reaction scheme involving a 1,5-hydrogen transfer followed by a cyclization step that finally leads to a new CâC bond formation could be verified. Transient absorption spectroscopy and DFT calculations trace convincingly the intermediates and transition states along the reaction path (see scheme).
In methods based on frozen-density embedding theory or subsystem formulation of density functional theory, the non-additive kinetic potential (vtnad(r)) needs to be approximated. Since vtnad(r) is defined as a bifunctional, the common strategies rely on approximating vtnad[ÏA,ÏB](r). In this work, the exact potentials (not bifunctionals) are constructed for chemically relevant pairs of electron densities (ÏA and ÏB) representing: dissociating molecules, two parts of a molecule linked by a covalent bond, or valence and core electrons. The method used is applicable only for particular case, where ÏA is a one-electron or spin-compensated two-electron density, for which the analytic relation between the density and potential exists. The sum ÏA + ÏB is, however, not limited to such restrictions. Kohn-Sham molecular densities are used for this purpose. The constructed potentials are analyzed to identify the properties which must be taken into account when constructing approximations to the corresponding bifunctional. It is comprehensively shown that the full von Weizsäcker component is indispensable in order to approximate adequately the non-additive kinetic potential for such pairs of densities.
Dye Bonding to TiO2: In Situ Attenuated Total Reflection Infrared Spectroscopy Study, Simulations, and Correlation with Dye-Sensitized Solar Cell Characteristics
B. Völker, F. Wölzl, T. Bürgi and D. Lingenfelser Langmuir, 28 (31) (2012), p11354-11363
DOI:10.1021/la302197z | unige:22919 | Abstract | Article HTML | Article PDF
Processing dye-sensitized solar cells gains more and more importance as interest in industrial applications grows daily. For large-scale processing and optimizing manufacturing in terms of environmental acceptability as well as time and material saving, a detailed knowledge of certain process steps is crucial. In this paper we concentrate on the sensitizing step of production, i.e., the anchoring of the dye molecules onto the TiO2 semiconductor. A vacuum-tight attentuated total reflection infrared (ATR-IR) flow-through cell was developed, thus allowing measurements using a vacuum spectrometer to monitor infiltration of dye molecules into the porous TiO2 film in situ at high sensitivity. In particular, the influence of the anchor and backbone of perylene dye molecules as well as the influence of solvents on the adsorption process was investigated. The experiments clearly show that an anhydride group reacts much slower than an acid group. A significantly lower amount of anhydride dye can be adsorbed on the films. Ex situ transmission experiments furthermore indicate that the availability of OH groups on the TiO2 surface may limit dye adsorption. Also the backbone and base frame of the dye can influence the adsorption time drastically. Electrical cell characteristics correlate with the amount of adsorbed dye molecules determined by in situ ATR-IR measurements. The latter is also sensitive toward the diffusion of the dye through the porous layer. To gain a deeper understanding of the interplay between diffusion and adsorption, simulations were performed that allowed us to extract diffusion and adsorption constants. Again it was demonstrated that the anchoring group has a strong effect on the adsorption rate. The influence of the solvent was also studied, and it was found that both adsorption and desorption are affected by the solvent. Protic polar solvents are able to remove bound dye molecules, which is a possible pathway of cell degradation. Most importantly, the analysis shows the potential of this approach for the evaluation of molecules or additives concerning their characteristics important for cell processing.
Thermal and Light-Induced Spin Switching Dynamics in the 2D Coordination Network of {[Zn1-xFex(bbtr)3](ClO4)2}∞: The Role of Cooperative Effects
P. Chakraborty, C. Enachescu, C. Walder, R. Bronisz and A. Hauser Inorganic Chemistry, 51 (18) (2012), p9714-9722
DOI:10.1021/ic301006c | unige:22920 | Abstract | Article HTML | Article PDF
The thermal spin transition, the photoexcitation, and the subsequent spin relaxation in the mixed crystal series of the covalently linked two-dimensional network {[Zn1-xFex(bbtr)3](ClO4)2}â (x = 0.02â1, bbtr =1,4-di(1,2,3-triazol-1-yl)-butane) are discussed. In the neat compound, the thermal spin transition with a hysteresis of 13 K is accompanied by a crystallographic phase transition (Kusz, J.; Bronisz, R.; Zubko, M.; Bednarek, H. Chem. Eur. J.2011, 17, 6807). In contrast, the diluted crystals with x ⤠0.1 stay essentially in the high-spin state down to low temperatures and show typical first order relaxation kinetics upon photoexcitation, and the structural phase transition is well separated from the spin transition. With increasing Fe(II) concentration, steeper thermal transitions and sigmoidal relaxation curves indicate increasingly important cooperative effects. Already at x = 0.38, the spin relaxation is governed by cooperative interactions between Fe(II) centers, and the crystallographic phase transition begins to influence the spin transition. The kinetic behavior of the thermal spin transition is reproduced within the framework of a dynamic mean-field model.
Liquid/liquid interfaces play a crucial role in numerous areas of science. However, direct spectroscopic access to this thin (â¼1 nm) region is not possible with conventional optical methods. After a brief review of the most used techniques to perform interfacial optical spectroscopy, we will focus on time-resolved surface second harmonic generation, which allows the measurement of the excited-state dynamics of probe molecules at interfaces. By comparing these dynamics with those measured in bulk solutions, precious information on the properties of the interfacial region can be obtained. To illustrate this, several studies performed in our group will be presented.
Revealing the Bonding Pattern from the Molecular Electron Density Using Single Exponential Decay Detector: An Orbital-Free Alternative to the Electron Localization Function
P. De Silva, J. Korchowiec and T.A. Wesolowski ChemPhysChem, 13 (15) (2012), p3462-3465 Keywords: bonding;computational chemistry;electron density;electron localization function;single exponential decay detector
DOI:10.1002/cphc.201200500 | unige:23744 | Abstract | Article PDF
We introduce a new tool (single exponential decay detector: SEDD) to extract information about bonding and localization in atoms, molecules, or molecular assemblies. The practical evaluation of SEDD does not require any explicit information about the orbitals. The only quantity needed is the electron density (calculated or experimental) and its derivatives up to the second order.
A Synthetic and Mechanistic Investigation of the Chromium Tricarbonyl-Mediated Masamune–Bergman Cyclization. Direct Observation of a Ground-State Triplet p-Benzyne Biradical
K.E.O. Ylijoki, S. Lavy, A. Fretzen, E.P. Kündig, T. Berclaz, G. Bernardinelli and C. Besnard Organometallics, 31 (15) (2012), p5396-5404
DOI:10.1021/om300427j | unige:22151 | Abstract | Article HTML | Article PDF
A new room-temperature chromium tricarbonyl-mediated cycloaromatization of enediynes is reported. The reaction occurs with both cyclic and acyclic enediynes in the presence of [Cr(CO)3(η6-naphthalene)] and both a coordinating solvent and a hydrogen atom source, providing chromiumâarene complexes in reasonable yield and good diastereocontrol. The mechanism of the reaction has been probed through DFT computational and spectroscopic methods. These studies suggest that direct C1âC6 bond formation from an η6-enediyne complex is the lowest-energy path, forming a metal-bound p-benzyne biradical. NMR spectroscopy suggests that enediyne alkene coordination occurs in preference to alkyne coordination, forming a THF-stabilized olefin intermediate; subsequent alkyne coordination leads to cyclization. While biradical quenching occurs rapidly and primarily via the singlet biradical, the triplet state biradical is detectable by EPR spectroscopy, suggesting intersystem crossing to a triplet ground state.
This work illustrates a simple approach for optimizing long-lived near-infrared lanthanide-centered luminescence using trivalent chromium chromophores as sensitizers. Reactions of the segmental ligand L2 with stoichiometric amounts of M(CF3SO3)2 (M = Cr, Zn) and Ln(CF3SO3)3 (Ln = Nd, Er, Yb) under aerobic conditions quantitatively yield the D3-symmetrical trinuclear [MLnM(L2)3](CF3SO3)n complexes (M = Zn, n = 7; M = Cr, n = 9), in which the central lanthanide activator is sandwiched between the two transition metal cations. Visible or NIR irradiation of the peripheral Cr(III) chromophores in [CrLnCr(L2)3]9+ induces rate-limiting intramolecular intermetallic CrâLn energy transfer processes (Ln = Nd, Er, Yb), which eventually produces lanthanide-centered near-infrared (NIR) or IR emission with apparent lifetimes within the millisecond range. As compared to the parent dinuclear complexes [CrLn(L1)3]6+, the connection of a second strong-field [CrN6] sensitizer in [CrLnCr(L2)3]9+ significantly enhances the emission intensity without perturbing the kinetic regime. This work opens novel exciting photophysical perspectives via the buildup of non-negligible population densities for the long-lived doubly excited state [Cr*LnCr*(L2)3]9+ under reasonable pumping powers.
Thiolate-protected gold nanoparticles and clusters combine size-dependent physical properties with the ability to introduce (bio)chemical functionality within their ligand shell. The engineering of the latter with molecular precision is an important prerequisite for future applications. A key question in this respect concerns the flexibility of the gold â sulfur interface. Here we report the first study on racemization of an intrinsically chiral gold nanocluster, Au38(SCH2CH2Ph)24, which goes along with a drastic rearrangement of its surface involving place exchange of several thiolates. This racemization takes place at modest temperatures (40 â 80 °C) without significant decomposition. The experimentally determined activation energy for the inversion reaction is ca 28 kcal/mol, which is surprisingly low considering the large rearrangement. The activation parameters furthermore indicate that the process occurs without complete Au-S bond breaking.
The effect of viscosity on the bimolecular electron transfer quenching of a series of coumarins by N,N-dimethylaniline was investigated using steady-state and time-resolved fluorescence spectroscopy. The data reveal that the static and transient stages of the quenching become dominant as viscosity increases. When extracting the quenching rate constants using a simple SternâVolmer analysis, a decrease of the rate constant with increasing driving force is observed above ~2 cP. However, this apparent Marcus inverted region, already reported several times with the same system in micelles and room temperature ionic liquids, totally vanishes when analyzing the data with a model accounting for the static and transient stages of the quenching. It appears that the apparent Marcus inverted region arises from the neglect of these quenching regimes together with the use of fluorophores with different excited-state lifetimes.
The excited-state dynamics of a series of Wursterâs salts (p-phenylenediamine radical cations) with different subtituents on the nitrogen atoms was investigated under a variety of experimental conditions using a combination of ultrafast spectroscopic techniques. At room temperature, the lifetime of the lowest excited state of all radical cations is on the order of 200 fs, independently of the solvent, that is, water, nitriles, alcohols, and room-temperature ionic liquid. On the other hand, all cations, except that with the bulky nitrogen substituents, become fluorescent below 120 K. The observed dynamics can be accounted for by the presence of a conical intersection between the D1Â and D0Â states. For the cations with a small nitrogen substituent, this conical intersection could be accessed through a twist of one amino group, as already suggested for Wursterâs Blue. However, this coordinate cannot be invoked for the cation with bulky nitrogen subtituents, and more probably, pyramidalization of the nitrogen center and/or deformation of the phenyl ring play an important role. Consequently, the excited-state dynamics of these structurally very similar Wursterâs salts involves different decay mechanisms.
The hydration of Th(IV) in ThCl4 and ThBr4 water solutions at different salt concentrations was studied in order to understand the structure of Th(IV) in liquid water and the effect of Brâ and Clâ anions on its hydration structure. Several theoretical methods were employed: density functional theory and classical molecular dynamics based on both semiempirical polarizable potentials and ab initio derived polarizable potentials. The results of the computations were combined with extended X-ray absorption fine structure (EXAFS) experimental data. The results of this study show that in pure water the ThâO distance of 2.45 Ã corresponds to a first shell coordination number between 9 and 10. In the salt solutions, while Brâ does not affect directly the hydration of Th(IV) also at relatively high concentrations, Clâ, on the other hand, is more structured around Th(IV), in agreement with recent high-energy X-ray scattering experiments. Counterions, even at relatively high concentrations (0.8 m), do not enter in the first solvation shell of Th(IV), but they induce an increase of water molecules in the first and second hydration shells of Th(IV).
Although liquid/liquid and air/liquid interfaces are omnipresent, very little is known up to now about the dynamics of processes occurring at such interfaces. As a detailed understanding of these processes could be of invaluable technological, environmental, and medical importance, considerable effort has been invested over the last two decades in developing new interface-selective techniques that allow for gaining further insight into the dynamics of these processes. Whereas several major results have been achieved that helped to contribute to a deeper understanding, there are still many aspects concerning the properties of liquid interfaces that are not yet fully understood. In this Perspective, the work that has been carried out so far on photoinduced interfacial dynamics will be reviewed and the current challenges in this still emerging field of research discussed.
Within the linear combination of atomic orbitals (LCAO) approximation, one can distinguish two different Kohn-Sham potentials. One is the potential available numerically in calculations, and the other is the exact potential corresponding to the LCAO density. The latter is usually not available, but can be obtained from the total density by a numerical inversion procedure or, as is done here, analytically using only one LCAO Kohn-Sham orbital. In the complete basis-set limit, the lowest-lying Kohn-Sham orbital suffices to perform the analytical inversion, and the two potentials differ by no more than a constant. The relation between these two potentials is investigated here for diatomic molecules and several atomic basis sets of increasing size and quality. The differences between the two potentials are usually qualitative (wrong behavior at nuclear cusps and far from the molecule even if Slater-type orbitals are used) and δ-like features at nodal planes of the lowest-lying LCAO Kohn-Sham orbital. Such nodes occur frequently in LCAO calculations and are not physical. Whereas the behavior of the potential can be systematically improved locally by the increase of the basis sets, the occurrence of nodes is not correlated with the size of the basis set. The presence of nodes in the lowest-lying LCAO orbital can be used to monitor whether the effective potential in LCAO Kohn-Sham equations can be interpreted as the potential needed for pure-state noninteracting v-representability of the LCAO density. Squares of such node-containing lowest-lying LCAO Kohn-Sham orbitals are nontrivial examples of two-electron densities which are not pure-state noninteracting v-representable.
Chirality unveiled: Thiolate-protected Au40(SR)24 clusters were enantioenriched using an HPLC approach. CD spectra show strong mirror-image responses, indicating the intrinsic chirality of a cluster of unknown structure protected with achiral ligands.
The potential of solid-phase microextraction on polyacrylate coated fibre, with sequential or simultaneous trimethylsilyl derivatisation followed by gas chromatographicâmass spectrometric analysis, was evaluated for a rapid determination of the distribution of the phytosterols in aqueous food matrixes. Influences of different parameters (bis(trimethylsilyl)trifluoro-acetamide and sterol exposure time, sterol concentration and experimental protocol) on the recovery of sterols were investigated to determine optimum conditions which were tested for sterol extraction and analysis from orange juice. Best selectivity, sterol recovery and derivatisation yields were obtained by extraction and simultaneous derivatisation through immersion of the SPME-PA fibre in the orange juice (10 min, 65 °C) after headspace absorption of BSTFA (30 min, 65 °C) on the fibre. Nevertheless the method developed cannot be used for quantitative analysis. But the possibility to effect rapid screen of phytosterol containing in complex media have been shown.
Silver nanoclusters protected by 2-phenylethanethiol (1), 4-fluorothiophenol (2), and l-glutathione (3) ligands were successfully synthesized. The optical properties of the prepared silver nanoclusters were studied. The absorption signal of Ag@SCH2CH2Ph in toluene can be found at 469 nm, and Ag@SPhF in THF shows two absorption bands at 395 and 462 nm. Ag@SG in water absorbs at 478 nm. Mie theory in combination with the Drude model clearly indicates the peaks in the spectra originate from plasmonic transitions. In addition, the damping constant as well as the dielectric constant of the surrounding medium was determined. In addition, the CD spectra of silver nanoclusters protected by the three ligands (1â3) were also studied. As expected, only the clusters of type 3 gave rise to chiroptical activity across the visible and near-ultraviolet regions. The location and strength of the optical activity suggest an electronic structure of the metal that is highly sensitive to the chiral environment imposed by the glutathione ligand. The morphology and size of the prepared nanoclusters were analyzed by using transmission electron microscopy (TEM). TEM analysis showed that the particles of all three types of silver clusters were small than 5 nm, with an average size of around 2 nm. The analysis of the FTIR spectra elucidated the structural properties of the ligands binding to the nanoclusters. By comparing the IR absorption spectra of pure ligands with those of the protected silver nanoclusters, the disappearance of the SâH vibrational band (2535â2564 cmâ1) in the protected silver nanoclusters confirmed the anchoring of ligands to the cluster surface through the sulfur atom. By elemental analysis and thermogravimetric analysis, the Ag/S ratio and, hence, the number of ligands surrounding a Ag atom could be determined.
Four novel bimetallic borohydrides have been discovered, K2M(BH4)4 (M = Mg or Mn), K3Mg(BH4)5, and KMn(BH4)3, and are carefully investigated structurally as well as regarding their decomposition reaction mechanism by means of in situ synchrotron radiation powder X-ray diffraction (SR-PXD), vibrational spectroscopies (Raman and IR), thermal analysis (TGA and DTA), and ab initio density functional theory (DFT) calculations. Mechano-chemical synthesis (ball-milling) using the reactants KBH4, α-Mg(BH4)2, and α-Mn(BH4)2 ensures chlorine-free reaction products. A detailed structural analysis reveals significant similarities as well as surprising differences among the two isomorphs K2M(BH4)4, most importantly concerning the extent to which the complex anion [M(BH4)4]2â is isolated in the structure. Anisotropic thermal expansion and an increase in symmetry at high temperatures in K3Mg(BH4)5 is ascribed to the motion of BH4 groups inducing hydrogen repulsive effects, and the dynamics of K3Mg(BH4)5 are investigated. Decomposition in the manganese system proceeds via the formation of KMn(BH4)3, the first perovkite type borohydride reported to date.
Ligand exchange reactions on size-selected Au38(2-PET)24 and Au40(2-PET)24 clusters (2-PET: 2-phenylethylthiol) with mono- and bidentate chiral thiols was performed. The reactions were monitored with MALDI mass spectrometry and the arising chiroptical properties were compared to the number of incorporated chiral ligands. Only a small fraction of chiral ligands is needed to induce significant optical activity to the clusters. The use of monodentate camphor-10-thiol (CamSH) leads to comparably fast exchange on both clusters. The arising optical activity is weak. In contrast, the use of bidentate 1,1â-binaphthyl-2,2â-dithiol (BINAS) is slow, but the optical activity measured is strong. Moreover, a non-linear behaviour between optical activity and number of chiral ligands is found in the BINAS case for both Au38 and Au40, which may indicate different exchange rates of enantiopure BINAS with the enantiomers of inherently chiral clusters. This is ascribed to effects arising from the bidentate nature of BINAS. This is the first study where chiroptical effects are directly correlated with the composition of the ligand shell.
First enantioseparation and circular dichroism spectra of Au38 clusters protected with achiral ligands
I. Dolamic, S. Knoppe, A. Dass and T. Bürgi Nature Communications, 3 (2012), p798
DOI:10.1038/ncomms1802 | unige:20260 | Abstract
Bestowing chirality to metals is central in fields such as heterogeneous catalysis and modern optics. Although the bulk phase of metals is symmetric, their surfaces can become chiral through adsorption of molecules. Interestingly, even achiral molecules can lead to locally chiral, though globally racemic, surfaces. A similar situation can be obtained for metal particles or clusters. Here we report the first separation of the enantiomers of a gold cluster protected by achiral thiolates, Au38(SCH2CH2Ph)24, achieved by chiral high-performance liquid chromatography. The chirality of the nanocluster arises from the chiral arrangement of the thiolates on its surface, forming 'staple motifs'. The enantiomers show mirror-image circular dichroism responses and large anisotropy factors of up to 4Ã10â3. Comparison with reported circular dichroism spectra of other Au38 clusters reveals that the influence of the ligand on the chiroptical properties is minor.
N-Aryl, N-branched alkyl carbamates react with in situ generated chiral Pd-NHC catalysts by coupling a Pd-Ar moiety with an aliphatic CâH bond at high temperature to give enantioenriched 2-substituted and 2,3-disubstituted indolines. Prochiral precursors give single products with very high asymmetric induction. Chiral racemic precursors react in a regiodivergent reaction of a racemic mixture to yield enantioenriched indolines resulting from either methyl CâH activation or asymmetric methylene CâH activation. In favorable cases this can result in a complete separation of an enatiomeric mixture into two different highly enantioenriched indolines.
Colloidal Mn2+-doped semiconductor nanocrystals such as Mn2+:ZnSe have attracted broad attention for potential applications in phosphor and imaging technologies. Here, we report saturation of the sensitized Mn2+ photoluminescence intensity at very low continuous-wave (CW) and quasi-CW photoexcitation powers under conditions that are relevant to many of the proposed applications. Time-resolved photoluminescence measurements and kinetic modeling indicate that this saturation arises from an Auger-type nonradiative cross relaxation between an excited Mn2+ ion and an exciton within the same nanocrystal. A lower limit of k = 2 Ã 1010 sâ1 is established for the fundamental rate constant of the Mn2+(4T1)-exciton cross relaxation.
Photoinduced Symmetry-Breaking Charge Separation
E. Vauthey ChemPhysChem, 13 (2012), p2001-2011 Keywords: excitation energy hopping;multichromophoric systems;photoinduced electron transfer;photochemistry;ultrafast spectroscopy
DOI:10.1002/cphc.201200106 | unige:21591 | Abstract | Article PDF
Molecular systems where several apparently equivalent charge separation pathways exist upon photoexcitation are presented. They encompass MQn (nâ¥2) architectures, where M is a chromophore and Q an electron transfer quencher (either donor or acceptor), and MâM systems where M acts as both electron donor and acceptor. In all cases, charge separation involves symmetry breaking. The conditions for such process to be operative as well as the origin of the symmetry breaking are discussed.
Hydrogen production from waste feedstocks using supercritical water gasification (SCWG) is a promising approach towards cleaner fuel production and a solution for hard to treat wastes. In this study, the catalytic co-gasification of starch and catechol as models of carbohydrates and phenol compounds was investigated in a batch reactor at 28 MPa, 400â500 °C, from 10 to 30 min. The effects of reaction conditions, and the addition of calcium oxide (CaO) as a carbon dioxide (CO2) sorbent and TiO2 as catalyst on the gas yields and product distribution were investigated. Employing TiO2 as a catalyst alone had no significant effect on the H2 yield but when combined with CaO increased the hydrogen yield by 35% and promoted higher total organic carbon (TOC) reduction efficiencies. The process liquid effluent was characterized using GCâMS, with the results showing that the major non-polar components were phenol, substituted phenols, and cresols. An overall reaction scheme is provided.
Ligand dependence of the synthetic approach and chiroptical properties of a magic cluster protected with a bicyclic thiolate
S. Knoppe, N. Kothalawala, V. Jupally, A. Dass and T. Bürgi ChemComm, 48 (2012), p4630-4632
DOI:10.1039/C2CC00056C | unige:20258 | Abstract | Article HTML | Article PDF
Chiral gold clusters stabilised by enantiopure thiolates were prepared, size-selected and characterised by Circular Dichroism and mass spectrometry. The product distribution is found to be ligand dependent. Au25 clusters protected with camphorthiol show clear resemblance of their chiroptical properties with their glutathionate analogue.
The photophysical properties of a series of helicene cations in various solvents have been investigated using stationary and time-resolved spectroscopy. These compounds fluoresce in the near infrared region with a quantum yield ranging between 2 and 20% and a lifetime between 1 and 12 ns, depending of the solvent. No clear solvent dependence could be recognized except for a decrease of fluorescence quantum yield and lifetime with increasing hydrogen-bond donating ability of the solvent. In water, the helicene cations undergo aggregation. This effect manifests itself by the presence of a slow fluorescence decay component, whose amplitude increases with dye concentration, and by a much slower decay of the polarization anisotropy in water compared to an organic solvent of similar viscosity. However, aggregation has essentially no effect on the stationary fluorescence spectrum, whereas relatively small changes can be seen in the absorption spectrum. Analysis of the dependence of aggregation on the dye concentration reveals that the aggregates are mostly dimers and that the aggregation constant is substantially larger for hetero- than homochiral dimers.
The influence of defects formed by Ca excess and thermal post-treatments on the persistent luminescence of CaTiO3:Pr
E.H. Otal, A.E. Maegli, N. Vogel-Schäuble, B. Walfort, H. Hagemann, S. Yoon, A. Zeller and A. Weidenkaff Optical Materials Express, 2 (4) (2012), p405-412
DOI:10.1364/OME.2.000405 | unige:18791 | Abstract | Article HTML | Article PDF
Red emitting CaTiO3:Pr phosphors with a nominal composition of Ca0.998+xPr0.002TiO3+δ (0.02â¤xâ¤0.04) were prepared by solid state reactions with different thermal post treatments and characterized by X-ray diffraction, transmission electron microscopy and photoluminescence. The Ca excess exhibited complete solubility up to 4% in the samples treated at 1400 °C but segregation in the form of Ruddlesden-Popper phases (Ca3Ti2O7 - Ca4Ti3O10) was observed in samples prepared at 1500 °C. The increase in temperature for stoichiometric samples showed a monotonic increase of decay time due to the reduction of non-radiative recombination defects. It was found that the Ca excess favored the formation of oxygen vacancies which are known to act as trap. In the samples treated at 1400 °C, 3% of Ca excess showed to be the best concentration to increase the decay time of persistent luminescence. For the samples treated at 1500 °C, the segregation of Ruddlesden-Popper phases left a constant amount of Ca soluble in all the CaTiO3 samples. This constant concentration of Ca caused the same density of defects and, consequently, the same decay time in all samples.
This contribution investigates LnIII complexes formed with a small ditopic ligand, L1, and their structural, thermodynamic and photophysical properties. The spectrophotometric and NMR titrations evidence the triangular assemblies [Ln3(L1-H)3]6+ at stoichiometric conditions and their properties are discussed in relation to L2-containing analogues. In addition, the dinuclear species, [Ln2(L1-H)]5+, is observed with an excess of metal.
Due to its extreme kinetic inertness, trivalent chromium, Cr(III), has been rarely combined with labile trivalent lanthanides, Ln(III), to give discrete self-assembled (supra)molecular polynuclear complexes. However, the plethora of accessible metal-centered excited states possessing variable lifetimes and emissive properties, combined with the design of efficient intramolecular Cr(III) â Ln(III) energy transfer processes open attractive perspectives for programming directional light-conversion within these heterometallic molecules. Efforts made to address this exciting challenge for both light-sensitization and light-upconversion are discussed in this article.
The crystal chemistry of the barium fluoride chloride system is studied both experimentally and theoretically. Different synthetic approaches yield nanocrystalline materials as well as large single crystals. The crystalline phases identified so far are BaFCl, Ba12F19Cl5 and Ba7F12Cl2 (in two modifications) and compared with analogous compounds. It is demonstrated that the compound Ba2F3Cl reported by Fessenden and Lewin 50 years ago corresponds to Ba7F12Cl2. The phase diagram of the BaCl2 â BaF2 system is reinvestigated for fluoride mole fractions between 0.5 and 1. The peritectic formation of Ba12F19Cl5 is observed. Periodic DFT calculations are performed for all structures in this system, including a hypothetical structure for Ba2F3Cl, based on the experimental structure of Ba2H3Cl. The energy of formation of the different barium fluoride chloride compounds from BaCl2 and BaF2 (normalized for one barium atom per formula unit), as calculated by DFT at 0K, is within only about ± 15 kJ/mol. Comparison with recent experimental results on calcium and strontium hydride chloride (bromide) compounds, suggest the possibility of a mutual exclusion between the M2X3Y and M7X12Y2 (M = Ca, Sr, Ba, Pb, X = H, F, Y = Cl,Br) structures. The single crystal structure of PbFBr is also reported.
In the covalently linked 2D coordination network {[Fe(bbtr)3](BF4)2}â, bbtr = 1,4-di(1,2,3-triazol-1-yl)butane, the iron(II) centers stay in the high-spin (HS) state down to 10 K. They can, however, be quantitatively converted to the low-spin (LS) state by irradiating into the near-IR spin allowed 5dd band and back again by irradiating into the visible 1dd band. The compound shows true light-induced bistability below 100 K, thus, having the potential for persistent bidirectional optical switching at elevated temperatures.
The fluorescence quenching of 3-cyanoperylene upon electron transfer from N,N-dimethylaniline in three room-temperature ionic liquids (RTILs) and in binary solvent mixtures of identical viscosity has been investigated using steady-state and time-resolved fluorescence spectroscopy. This study was stimulated by previous reports of bimolecular electron transfer reactions faster by one or several orders of magnitude in RTILs than in conventional polar solvents. These conclusions were usually based on a comparison with data obtained in low-viscous organic solvents and extrapolated to higher viscosities and not by performing experiments at similar viscosities as those of the RTILs, which we show to be essential. Our results reveal that (i) the diffusive motion of solutes in both types of solvents is comparable, (ii) the intrinsic electron transfer step is controlled by the solvent dynamics in both cases, being slower in the RTILs than in the conventional organic solvent of similar viscosity, and (iii) the previously reported reaction rates much larger than the diffusion limit at low quencher concentration in RTILs originate from a neglect of the static and transient stages of the quenching, which are dominant in solvents as viscous as RTILs.
NMR Study of Reorientational Motion in Alkaline-Earth Borohydrides: β and γ Phases of Mg(BH4)2 and α and β Phases of Ca(BH4)2
A.V. Soloninin, O.A. Babanova, A.V. Skripov, H. Hagemann, B. Richter, T.R. Jensen and Y. Filinchuk Journal of Physical Chemistry C, 116 (7) (2012), p4913-4920
DOI:10.1021/jp210509g | unige:18736 | Abstract | Article HTML | Article PDF
To study the reorientational motion of BH4 groups in β and γ phases of Mg(BH4)2 and in α and β phases of Ca(BH4)2, we have performed nuclear magnetic resonance (NMR) measurements of the 1H and 11B spinâlattice relaxation rates in these compounds over wide ranges of temperature and resonance frequency. It is found that at low temperatures the reorientational motion in β phases of Mg(BH4)2 and Ca(BH4)2 is considerably faster than in other studied phases of these alkaline-earth borohydrides. The behavior of the measured spinâlattice relaxation rates in both β phases can be satisfactorily described in terms of a Gaussian distribution of activation energies Ea with the average Ea values of 138 meV for β-Mg(BH4)2and 116 meV for β-Ca(BH4)2. The α phase of Ca(BH4)2 is characterized by the activation energy of 286 ± 7 meV. For the novel porous γ phase of Mg(BH4)2, the main reorientational process responsible for the observed spinâlattice relaxation rate maximum can be described by the activation energy of 276 ± 5 meV. The barriers for reorientational motion in different phases of alkaline-earth borohydrides are discussed on the basis of changes in the local environment of BH4 groups.
Computational Insights into Uranium Complexes Supported by Redox-Active α-Diimine Ligands
G. Li Manni, J.R. Walensky, S.J. Kraft, W.P. Forrest, L.M. Pérez, M.B. Hall, L. Gagliardi and S.C. Bart Inorganic Chemistry, 51 (4) (2012), p2058-2064
DOI:10.1021/ic202522w | unige:18735 | Abstract | Article HTML | Article PDF
The electronic structures of two uranium compounds supported by redox-active α-diimine ligands, (MesDABMe)2U(THF) (1) and Cp2U(MesDABMe) (2) (MesDABMe = [ArNâC(Me)C(Me)âNAr]; Ar = 2,4,6-trimethylphenyl (Mes)), have been investigated using both density functional theory and multiconfigurational self-consistent field methods. Results from these studies have established that both uranium centers are tetravalent, that the ligands are reduced by two electrons, and that the ground states of these molecules are triplets. Energetically low-lying singlet states are accessible, and some transitions to these states are visible in the electronic absorption spectrum.
Photophysical Properties of {[Au(CN)2]−}2 Dimers Trapped in a Supramolecular Electron-Acceptor Organic Framework
A.S. Abouelwafa, C.E. Anson, A. Hauser, H.H. Patterson, F. Baril-Robert, X. Li and A.K. Powell Inorganic Chemistry, 51 (3) (2012), p1294-1301
DOI:10.1021/ic201109u | unige:18374 | Abstract | Article HTML | Article PDF
Dicyanoaurate reacts with the organic acceptor molecule, 1,1â²-bis-(2,4-dinitrophenyl)-4,4â²-bipyridinium, DNP, to form a supramolecular complex with the general formula {[Au(CN)2]2DNP}·4H2O. The complex was characterized by X-ray crystallography, and its photophysical properties were investigated in the solid-state. Although the initial (DNP)Cl2 compound does not show photoluminescence behavior and the dicyanoaurate shows photoluminescence only in the UV range, the resulting supramolecular complex displays two simultaneous, essentially independent, photoluminescence bands in the visible range originating from individual contributions of the DNP unit and the dicyanoaurate dimers. This unusual simultaneous photoluminescence behavior displayed by both the dicyanoaurate donor units and the redox-active 4,4â²-bipyridinium acceptor have lifetimes of 0.5 μs and several hundred μs, respectively.
The photophysical properties of the free neutral radical galvinoxyl were studied by a combination of femtosecond time-resolved spectroscopy and quantum chemical calculations. The electronic absorption spectrum is dominated by an intense band at 430 nm that is ascribed to the D9,10âD0 transitions. Upon photoexcitation at 400 nm, the population of the D9,10 states decays within less than 200 fs to the electronic ground state. This ultrafast internal conversion does not involve intramolecular modes with large amplitude motion as the measured dynamics does not show any significant dependence on the environment, but is most probably facilitated by a high density of electronic states of different character. Depending on the solvent, a weak transient band due to the galvinoxylate anion is also observed. This closed-shell species, which is fluorescent although its deactivation is also dominated by non-radiative decay, is generated upon biphotonic ionization of the solvent and electron capture. The ultrashort excited-state lifetime of the galvinoxyl radical precludes photoinduced disproportionation previously claimed to be at the origin of the formation of both anion and cation.
Assessing Metal-Metal Multiple Bonds in Cr—Cr, Mo—Mo, and W—W Compounds and a Hypothetical U—U Compound: A Quantum Chemical Study Comparing DFT and Multireference Methods
G. Li Manni, A.L. Dzubak, A. Mulla, D.W. Brogden, J.F. Berry and L. Gagliardi Chemistry - A European Journal, 18 (6) (2012), p1737-1749 Keywords: bond theory;density functional calculations;electronic states;metal|metal interactions;quantum chemistry
DOI:10.1002/chem.201103096 | unige:18373 | Abstract | Article PDF
To gain insights into the trends in metalâmetal multiple bonding among the Groupâ 6 elements, density functional theory has been employed in combination with multiconfigurational methods (CASSCF and CASPT2) to investigate a selection of bimetallic, multiply bonded compounds. For the compound [Ar-MM-Ar] (Ar=2,6-(C6H5)2-C6H3, M=Cr, Mo, W) the effect of the Ar ligand on the M2 core has been compared with the analogous [Ph-MM-Ph] (Ph=phenyl, M=Cr, Mo, W) compounds. A set of [M2(dpa)4] (dpa=2,2â²-dipyridylamide, M=Cr, Mo, W, U) compounds has also been investigated. All of the compounds studied here show important multiconfigurational behavior. For the Mo2 and W2 compounds, the Ï2Ï4δ2 configuration dominates the ground-state wavefunction, contributing at least 75â%. The Cr2 compounds show a more nuanced electronic structure, with many configurations contributing to the ground state. For the Cr, Mo, and W compounds the electronic absorption spectra have been studied, combining density functional theory and multireference methods to make absorption feature assignments. In all cases, the main features observed in the visible spectra may be assigned as charge-transfer bands. For all compounds investigated the Mayer bond order (MBO) and the effective bond order (EBO) were calculated by density functional theory and CASSCF methods, respectively. The MBO and EBO values share a similar trend toward higher values at shorter normalized metalâmetal bond lengths.
Shifts in Excitation Energies Induced by Hydrogen Bonding: A Comparison of the Embedding and Supermolecular Time-Dependent Density Functional Theory Calculations with the Equation-of-Motion Coupled-Cluster Results
G. Fradelos, J.J. Lutz, T.A. Wesolowski, P. Piecuch and M. Wloch
in "Progress in Theoretical Chemistry and Physics"
Advances in the Theory of Quantum Systems in Chemistry and Physics, ed. P. Hoggan, E. Brändas, J. Maruani, P. Piecuch and G. Delgado-Barrio,
22 (2012), p219-248
DOI:10.1007/978-94-007-2076-3_13 | unige:17800 | Abstract | Article PDF
Shifts in the Ï â Ïâ excitation energy of the cis-7-hydroxyquinoline chromophore induced by hydrogen bonding with small molecules, obtained with the frozen-density embedding theory (FDET), are compared with the results of the high-level equation-of-motion coupled-cluster (EOMCC) calculations with singles, doubles, and noniterative triples, which provide the reference ab initio data, the supermolecular time-dependent density functional theory (TDDFT) calculations, and the available experimental data. It is demonstrated that the spectral shifts resulting from the FDET calculations employing nonrelaxed environment densities and their EOMCC counterparts are in excellent agreement with one another, whereas the analogous shifts obtained with the supermolecular TDDFT approach do not agree with the EOMCC reference data. Among the discussed issues are the effects of higher-order correlations on the excitation energies and complexation-induced excitation energy shifts resulting from the EOMCC calculations, and the choice of the approximants that represent the nonadditive kinetic energy contributions to the embedding potential of FDET.
Attenuated total reflection infrared (ATR-IR) spectroscopy, modulation excitation spectroscopy (MES), and vibrational circular dichroism (VCD)
T. Bürgi
in "Biointerface Characterization by Advanced IR Spectroscopy"
Edited by:C.-M. Pradier And Y.J. Chabal,
Ch. 5 (2011), p115-144
DOI:10.1016/B978-0-444-53558-0.00005-9 | unige:98056 | Abstract
This chapter describes the potential and limitations of attenuated total reflection infrared (ATR-IR) spectroscopy, modulation excitation spectroscopy (MES), and vibrational circular dichroism (VCD) for the investigation of bio-interfaces. The three techniques are completely independent of each other although ATR-IR and MES can be combined. ATR-IR is based on the total internal reflection of a light beam at an interface formed between two media. The resulting field at the interface is used for spectroscopy. The evanescent nature of this field allows one to probe the layer close to the interface formed between the internal reflection element (IRE) and the sample. Therefore, the field probes only a small part of the bulk fluid phase, such that molecules at the interface can be studied even in the presence of strongly absorbing solvents such as water. This makes the technique particularly interesting for applications in biology, where water is the solvent. The electric field can furthermore be polarized, which can be used for orientation measurements. MES is based on the stimulation of the sample under investigation by a periodic change of an external parameter such as temperature, pressure, pH, or concentration. The periodic system response to this stimulation (excitation) can be followed using a spectroscopic technique like ATR-IR by recording time-resolved spectra. The latter are then transformed into phase-resolved spectra using a phase-sensitive detection (PSD) scheme. MES leads to a drastic suppression of noise, which is very welcome when studying molecules at interfaces by spectroscopic techniques. Furthermore, MES helps to disentangle complex overlapping spectra arising from several species.
Optical properties of a fabricated self-assembled bottom-up bulk metamaterial
S. Mühlig, C. Rockstuhl, V. Yannopapas, T. Bürgi, N. Shalkevich and F. Lederer Optics Express, 19 (10) (2011), p9607-9616
DOI:10.1364/OE.19.009607 | unige:98050 | Abstract | Article HTML | Article PDF
We investigate the optical properties of a true three-dimensional metamaterial that was fabricated using a self-assembly bottom-up technology. The metamaterial consists of closely packed spherical clusters being formed by a large number of non-touching gold nanoparticles. After presenting experimental results, we apply a generalized Mie theory to analyze its spectral response revealing that it is dominated by a magnetic dipole contribution. By using an effective medium theory we show that the fabricated metamaterial exhibits a dispersive effective permeability, i.e. artificial magnetism. Although this metamaterial is not yet left-handed it might serve as a starting point for achieving bulk metamaterials by using bottom-up approaches.
The self-assembly of core-substituted naphthalene diimides bearing triethylene glycol motifs leads to the formation of stable vesicles in DMSO and CHCl3/MeOH (6 : 4, v/v) solvents. The vesicles were evaluated by means of UV/vis and fluorescence spectroscopy, transmission electron microscopy, atomic force microscopy and dynamic light scattering.
Crystal growth and structure determination of the novel tetragonal compound Ce2RhGa12
R. Nagalakshmi, R. Kulkarni, S.K. Dhar, A. Thamizhavel, V. Krishnakumar, C. Besnard, H. Hagemann and M. Reiffers Chemistry of metals and alloys, 4 (3/4) (2011), p229-233 Keywords: Crystal structure, intermetallic compound
unige:23118 | Abstract | Article PDF
Single crystals of Ce2RhGa12 have been synthesized using Ga flux and their structure was determined by single-crystal X-ray diffraction. Ce2RhGa12 crystallizes in the tetragonal space group P4/nbm (No. 125), and is isostructural to Ce2PdGa12, with Z = 2 and lattice parameters a = 6.0405 à and c = 15.706 à . Data were collected at the Swiss Norwegian Beam Line at the European Synchrotron Facility, Grenoble, France. Laue diffraction was carried out to confirm the quality of the single crystal and showed well-defined spots and tetragonal symmetry.
The green-fluorescent protein of the jellyfish operates with the most powerful phenolate donors in the pushâpull fluorophore. To nevertheless achieve red fluorescence with the same architecture, sea anemone and corals apply oxidative imination, a process that accounts for the chemistry of vision as well. The objective of this study was to apply these lessons from nature to one of the most compact family of panchromatic fluorophores, i.e. core-substituted naphthalenediimides (cNDIs). We report straightforward synthetic access to hydroxylated cNDI and cPDI cores by palladium-catalyzed cleavage of allyloxy substituents. With hydroxylated cNDIs but not cPDIs in water-containing media, excited-state intramolecular proton transfer yields a second bathochromic emission. Deprotonation of hydroquinone, catechol and boronic ester cores provides access to an impressive panchromism up to the NIR frontier at 640 nm. With cNDIs, oxidative imination gives red shifts up to 638 nm, whereas the expanded cPDIs already absorb at 754 nm upon deprotonation of hydroquinone cores. The practical usefulness of hydroquinone cNDIs is exemplified by ratiometric sensing of the purity of DMF with the ânaked eyeâ at a sensitivity far beyond the ânaked noseâ. We conclude that the panchromatic hypersensitivity toward the environment of the new cNDIs is ideal for pattern generation in differential sensing arrays.
Modified ene-yne compounds: a novel functional material with nonlinear optical properties
D. Lumpi, B. Stöger, C. Hametner, F. Kubel, G. Reider, H. Hagemann, A. Karpfen and J. Fröhlich CrystEngComm, 13 (24) (2011), p7194-7197
DOI:10.1039/C1CE06093G | unige:17799 | Article HTML | Article PDF
The title compound, an achiral flexible molecule containing a 1,2,3-triazole structure as the acceptor subunit, crystallizes as a single enantiomorph in the space group P212121. The material exhibits nonlinear optical properties and is capable of second harmonic generation. Thus, the developed molecular scaffold represents an interesting novel type of NLO chromophore.
Two molecules containing two phenylphosphaalkene moieties linked by an anthracene (1) or by a naphthalene (2) ring have been synthesized and their crystal structures have been determined. While electrochemical measurements show that these two systems are easily reduced, EPR spectra indicate that, at room temperature, the electronic structures of the two reduction compounds 1Ëâ and 2Ëâ are quite different. In 1Ëâ, in good accordance with DFT predictions, the unpaired electron is delocalized on the full molecule while in 2Ëâ it is confined on a single phosphaalkene moiety. This difference is attributed to the short distance between the two phenylphosphaalkene groups in 2Ëâ which hinders their reorientation after addition of an electron. The role of this motion is consistent with the fact that two additional paramagnetic species are detected at 145 K: the dianion 22â characterized by a rather small exchange coupling constant and the radical monoanion 2*Ëâ resulting from the formation of a one electron PâP bond. These two species are probably reaction intermediates which can lead to the formation of biphosphane.
Thermal hysteresis in spin-crossover compounds studied within the mechanoelastic model and its potential application to nanoparticles
C. Enachescu, P. Chakraborty, L. Stoleriu, A. Stancu and A. Hauser Physical Review B, 84 (13) (2011), p134102
DOI:10.1103/PhysRevB.84.134102 | unige:17482 | Abstract | Article PDF
The recently developed mechanoelastic model is applied to characterize the thermal transition in spin-crossover complexes, with special attention given to the case of spin-crossover nanoparticles. In a two-dimensional system, hexagonal-shaped samples with open boundary conditions are composed of individual molecules that are linked by springs and can switch between two states, namely, the high-spin (HS) and the low-spin (LS) states. The switching of an individual molecule during the spin transition is decided by way of a Monte Carlo standard procedure, using transition probabilities depending on the temperature, the energy gap between the two states, the enthalpy difference, the degeneracy ratio, and the local pressure determined by the elongation or compression of its closest springs. The influence of external parameters, such as temperature sweeping rate and pressure, or intrinsic features of the system, such as the value of its spring constant, on the width of the thermal hysteresis, its shape, and its position are discussed. The particular case of spin-crossover nanoparticles is treated by considering them embedded into a polymer environment, which essentially affects the molecules situated at the edges and faces by decreasing their transition probabilities from HS to LS. Finally, the pressure hysteresis, obtained by varying the external pressure at constant temperature is discussed.
Targeting π-Conjugated Multiple Donor–Acceptor Motifs Exemplified by Tetrathiafulvalene-Linked Quinoxalines and Tetrabenz[bc,ef,hi,uv]ovalenes: Synthesis, Spectroscopic, Electrochemical, and Theoretical Characterization
H.-P. Jia, J. Ding, Y.-F. Ran, S.-X. Liu, C. Blum, I. Petkova, A. Hauser and S. Decurtins Chemistry - An Asian Journal, 6 (12) (2011), p3312-3321 Keywords: charge transfer;donor-acceptor systems;nanographene;redox chemistry;tetrathiafulvalene
DOI:10.1002/asia.201100515 | unige:18010 | Abstract | Article PDF
An efficient synthetic approach to a symmetrically functionalized tetrathiafulvalene (TTF) derivative with two diamine moieties, 2-[5,6-diamino-4,7-bis(4-pentylphenoxy)-1,3-benzodithiol-2-ylidene]-4,7-bis(4-pentylphenoxy)-1,3-benzodithiole-5,6-diamine (2), is reported. The subsequent Schiff-base reactions of 2 afford large Ï-conjugated multiple DâA arrays, for example the triad 2-[4,9-bis(4-pentylphenoxy)-1,3-dithiolo[4,5-g]quinoxalin-2-ylidene]-4,9-bis(4-pentylphenoxy)-1,3-dithiolo[4,5-g]quinoxaline (8) and the corresponding tetrabenz[bc,ef,hi,uv]ovalene-fused pentad 1, in good yields and high purity. The novel redox-active nanographene 1 is so far the largest known TTF-functionalized polycyclic aromatic hydrocarbon with a well-resolved 1H NMR spectrum. The electrochemically highly amphoteric pentad 1 and triad 8 exhibit various electronically excited charge-transfer states in different oxidation states leading to intense optical intramolecular charge transfer (ICT) absorbances over a wide spectral range. The chemical and electrochemical oxidations of 1 result in an unprecedented TTFâ¢+ radical cation dimerization, leading to the formation of [1â¢+]2 at room temperature in solution due to the stabilizing effect arising from strong ÏâÏ interactions. Moreover, ICT fluorescence is observed with large solvent-dependent Stokes shifts and quantum efficiencies of 0.05 for 1 and 0.035 for 8 in CH2Cl2.
Study of the relaxation in diluted spin crossover molecular magnets in the framework of the mechano-elastic model
C. Enachescu, L. Stoleriu, A. Stancu and A. Hauser Journal of Applied Physics, 109 (7) (2011), p711 Keywords: magnetic impurities, metastable states, molecular magnetism, Monte Carlo methods, spin dynamics
DOI:10.1063/1.3556702 | unige:17481 | Abstract | Article HTML | Article PDF
We model here the behavior of spin transition compounds, considering molecules arranged in a 2D hexagonal lattice and interacting via springs. The role of impurities in the clustering and nucleation phenomena is analyzed, as well as the manner in which the impurities affect the relaxation curves. The switching of the individual molecules is checked using a Monte Carlo procedure. When a molecule changes its state, it also modifies its volume, and the new equilibrium positions of all the molecules are calculated. As in previously reported experiments, it is found here that bigger impurities slow down the relaxation from the metastable high-spin state to the low-spin state, while smaller impurities act in an opposite way. It is shown that if the concentration of the impurities is higher than a certain threshold, then they act as a barrier, trammeling the fast evolution of domains developing from the edges.
PURPOSE. To compare the photochemical properties of UV filter molecules present in the human lens (kynurenine, KN; 3-hy- droxykynurenine, 3OHKN; 3-hydroxykynurenine O-gluco- side, 3OHKG; 4-(2-aminophenyl)-4-oxobutanoic acid, AHA; and glutathionyl-kynurenine, GSH-KN) with the use of the following parameters: excited singlet lifetime, fluorescence quantum yield, triplet quantum yield, photodecomposition quantum yield.
METHODS. The excited singlet lifetimes were measured with the use of fluorescence upconversion (time resolution, 210 fs) and pump-probe transient absorption (time resolution, 200 fs) methods. The fluorescence quantum yields were determined relative to an aqueous solution of quinine bisulfate. The triplet quantum yields were measured with the use of nanosecond laser flash photolysis. The photodecomposition quantum yields were determined by steady state photolysis followed by the high-performance liquid chromatography analysis.
RESULTS. The secondary UV filtersâAHA and GSH-KN are better photosensitizers than the primary ones -KN, 3OHKN and 3OHKG: the singlet state lifetimes of the secondary UV filters are longer, and the quantum yields of fluorescence and triplet state formation are higher.
CONCLUSIONS. With aging, the ratio primary/secondary UV filters in the human lens decreases from approximately 10:1 to 2:1. The obtained results demonstrate that the quality of the secondary UV filters is inferior compared to the primary ones, which may result in a higher susceptibility of old lenses to UV light. That might be an important factor for the development of the age-related cataract.
Facile access to complex systems is crucial to generate the functional materials of the future. Herein, we report self-organizing surface-initiated polymerization (SOSIP) as a user-friendly method to create ordered as well as oriented functional systems on transparent oxide surfaces. In SOSIP, self-organization of monomers and ring-opening disulfide exchange polymerization are combined to ensure the controlled growth of the polymer from the surface. This approach provides rapid access to thick films with smooth, reactivatable surfaces and long-range order with few defects and high precision, including panchromatic photosystems with oriented four-component redox gradients. The activity of SOSIP architectures is clearly better than that of disordered controls.
Porous and Dense Magnesium Borohydride Frameworks: Synthesis, Stability, and Reversible Absorption of Guest Species
Y. Filinchuk, B. Richter, T.R. Jensen, V. Dmitriev, D. Chernyshov and H. Hagemann Angewandte Chemie International Edition, 50 (47) (2011), p11162-11166 Keywords: framework materials;host-guest systems;hydrides;hydrogen storage;polymorphism
DOI:10.1002/anie.201100675 | unige:17480 | Abstract | Article PDF
Highly occupied: A highly porous form of Mg(BH4)2Â (see picture; Mg green, BH4Â blue, unit cells shown in red) reversibly absorbs H2, N2, and CH2Cl2. At high pressures, this material transforms into an interpenetrated framework that has 79â% higher density than the other polymorphs. Mg(BH4)2Â can act as a coordination polymer that has many similarities to metalâorganic frameworks.
In this article, the synthesis of a novel high-conjugated ligand and its corresponding Ru(II) complex PTFTF:Ru is reported, along with the linear and nonlinear optical characterizations. Two-photon absorption based optical power limiting properties (OPL), especially in the near infrared, are described and compared to those of the analogous complexes previously published. Combined with a preliminary theoretical approach, this allows us to highlight several key parameters for OPL optimization in such molecular systems and more particularly the spectral overlap between TPA and excited-state absorption.
This work illustrates a simple approach for optimizing the lanthanide luminescence in molecular dinuclear lanthanide complexes and identifies a particular multidentate europium complex as the best candidate for further incorporation into polymeric materials. The central phenyl ring in the bis-tridentate model ligands L3âL5, which are substituted with neutral (X = H, L3), electron-withdrawing (X = F, L4), or electron-donating (X = OCH3, L5) groups, separates the 2,6-bis(benzimidazol-2-yl)pyridine binding units of linear oligomeric multi-tridentate ligand strands that are designed for the complexation of luminescent trivalent lanthanides, Ln(III). Reactions of L3âL5 with [Ln(hfac)3(diglyme)] (hfacâ is the hexafluoroacetylacetonate anion) produce saturated single-stranded dumbbell-shaped complexes [Ln2(Lk)(hfac)6] (k = 3â5), in which the lanthanide ions of the two nine-coordinate neutral [N3Ln(hfac)3] units are separated by 12â14 à . The thermodynamic affinities of [Ln(hfac)3] for the tridentate binding sites in L3âL5 are average (6.6 ⤠log(β2,1Y,Lk) ⤠8.4) but still result in 15â30% dissociation at millimolar concentrations in acetonitrile. In addition to the empirical solubility trend found in organic solvents (L4 > L3 >> L5), which suggests that the 1,4-difluorophenyl spacer in L4 is preferable, we have developed a novel tool for deciphering the photophysical sensitization processes operating in [Eu2(Lk)(hfac)6]. A simple interpretation of the complete set of rate constants characterizing the energy migration mechanisms provides straightforward objective criteria for the selection of [Eu2(L4)(hfac)6] as the most promising building block.
Thermodynamics, Structure and Properties of Polynuclear Lanthanide Complexes with a Tripodal Ligand: Insight into their Self-Assembly
J. Hamacek, C. Besnard, T. Penhouet and Chemistry - A European Journal, 17 (24) (2011), p6753-6764 Keywords: helical structures;lanthanides;polynuclear complexes;thermodynamics;tripodal ligands
DOI:10.1002/chem.201100173 | unige:17236 | Abstract | Article HTML | Article PDF
Self-assembly processes between a tripodal ligand and LnIIIÂ cations have been investigated by means of supramolecular analytical methods. At an equimolar ratio of components, tetranuclear tetrahedral complexes are readily formed in acetonitrile. The structural analysis of the crystallographic data shows a helical wrapping of binding strands around metallic cations. The properties of this series of highly charged 3D compounds were examined by using NMR spectroscopy and optical methods in solution and in the solid state. In the presence of excess metal, a new trinuclear complex was identified. The X-ray crystal structure elucidated the coordination of metallic cations with two ligands of different conformations. By varying the metal/ligand ratio, a global speciation of this supramolecular system has been evidenced with different spectroscopic methods. In addition, these rather complicated equilibria were successfully characterised with the thermodynamic stability constants. A rational analysis of the self-assembly processes was attempted by using the thermodynamic free energy model and the impact of the ligand structure on the effective concentration is discussed.
The structure and thermodynamic properties of lanthanide complexes with a new tripodal ligand L2 have been elucidated using different physicochemical methods. At stoichiometric ratios, the tetrahedral three-dimensional complexes with lanthanide cations are formed in acetonitrile with good stabilities. Despite minor structural changes comparing to previously investigated tripodal ligands, the resulting assembly exhibits different features revealed with the crystal structure of [Eu4L24](OH)(ClO4)11 (orthorhombic, Pbcn). Interestingly, the highly charged edifice contains an inner cage encapsulating a perchlorate anion. Such lanthanide mediated cage-like assemblies are rare, and may be of interest for different sensing applications. Indeed, the anionic guest can be exchanged with different anions. The related hostâguest equilibria were investigated with NMR techniques. Various aspects of these reactions are qualitatively discussed.
The bi-functional for the non-electrostatic part of the exact embedding potential of frozen-density embedding theory (FDET) depends on whether the embedded part is described by means of a real interacting many-electron system or the reference system of non-interacting electrons (see [Wesolowski, Phys. Rev. A. 77, 11444 (2008)]). The difference δÎFMD[ÏA] / δÏA(r), where ÎFMD[ÏA] is the functional bound from below by the correlation functional Ec[ÏA] and from above by zero. Taking into account ÎFMD[ÏA] in both the embedding potential and in energy is indispensable for assuring that all calculated quantities are self-consistent and that FDET leads to the exact energy and density in the limit of exact functionals. Since not much is known about good approximations for ÎFMD[ÏA], we examine numerically the adequacy of neglecting ÎFMD[ÏA] entirely. To this end, we analyze the significance of δÎFMD[ÏA] / δÏA(r) in the case where the magnitude of ÎFMD[ÏA] is the largest, i.e., for Hartree-Fock wavefunction. In hydrogen bonded model systems, neglecting δÎFMD[ÏA] / δÏ(r) in the embedding potential marginally affects the total energy (less than 5% change in the interaction energy) but results in qualitative changes in the calculated hydrogen-bonding induced shifts of the orbital energies. Based on this estimation, we conclude that neglecting δÎFMD[ÏA] / δÏA(r) may represent a good approximation for multi-reference variational methods using adequate choice for the active space. Doing the same for single-reference perturbative methods is not recommended. Not only it leads to violation of self-consistency but might result in large effect on orbital energies. It is shown also that the errors in total energy due to neglecting δÎFMD[ÏA] / δÏA(r) do not cancel but rather add up to the errors due to approximation for the bi-functional of the non-additive kinetic potential.
In embedding methods such as those labeled commonly as QM/MM, the embedding operator is frequently approximated by the electrostatic potential generated by nuclei and electrons in the environment. Such approximation is especially useful in studies of the potential energy surface of embedded species. The effect on energy of neglecting the non-Coulombic component of the embedding operator is corrected a posteriori. The present work investigates applicability of such approximation in evaluation of electronic excitation energy, the accuracy of which depends directly on that of the embedding potential. For several model systems involving cis-7-hydroxiquinoline hydrogen-bonded to small molecules, we demonstrate that such truncation of the embedding operator leads to numerically unstable results upon increasing the size of the atomic basis sets. Approximating the non-Coulombic component of the embedding potential using the expression derived in Frozen-Density Embedding Theory ([Wesolowski and Warshel, J. Phys. Chem.1993, 97, 8050] and subsequent works) by means of even a simple bifunctional dependent on the electron density of the chromophore and its hydrogen-bonded environment, restores the numerical stability of the excitation energies that reach a physically meaningful limit for large basis sets.
The generalized active space concept in multiconfigurational self-consistent field methods
D. Ma, G. Li Manni and L. Gagliardi Journal of Chemical Physics, 135 (4) (2011), p44128 Keywords: configuration interactions, gadolinium, manganese compounds, organic compounds, SCF calculations, wave functions
DOI:10.1063/1.3611401 | unige:16781 | Abstract | Article HTML | Article PDF
A multiconfigurational self-consistent field method based on the concept of generalized active space (GAS) is presented. GAS wave functions are obtained by defining an arbitrary number of active spaces with arbitrary occupation constraints. By a suitable choice of the GAS spaces, numerous ineffective configurations present in a large complete active space (CAS) can be removed, while keeping the important ones in the CI space. As a consequence, the GAS self-consistent field approach retains the accuracy of the CAS self-consistent field (CASSCF) ansatz and, at the same time, can deal with larger active spaces, which would be unaffordable at the CASSCF level. Test calculations on the Gd atom, Gd2 molecule, and oxoMn(salen) complex are presented. They show that GAS wave functions achieve the same accuracy as CAS wave functions on systems that would be prohibitive at the CAS level.
We present the femtosecond spectroscopic investigation of a covalently linked dyad, PCB-P3HT, formed by a segment of the conjugated polymer P3HT (regioregular poly(3-hexylthiophene)) that is end capped with the fullerene derivative PCB ([6,6]-phenyl-C61-butyric acid ester), adapted from PCBM. The fluorescence of the P3HT segment in tetrahydrofuran (THF) solution is reduced by 64% in the dyad compared to a control compound without attached fullerene (P3HT-OH). Fluorescence upconversion measurements reveal that the partial fluorescence quenching of PCB-P3HT in THF is multiphasic and occurs on an average time scale of 100 ps, in parallel to excited-state relaxation processes. Judging from ultrafast transient absorption experiments, the origin of the quenching is excitation energy transfer from the P3HT donor to the PCB acceptor. Due to the much higher solubility of P3HT compared to PCB in THF, the PCB-P3HT dyad molecules self-assemble into micelles. When pure C60 is added to the solution, it is incorporated into the fullerene-rich center of the micelles. This dramatically increases the solubility of C60 but does not lead to significant additional quenching of the P3HT fluorescence by the C60 contained in the micelles. In PCB-P3HT thin films drop-cast from THF, the micelle structure is conserved. In contrast to solution, quantitative and ultrafast (<150 fs) charge separation occurs in the solid-state films and leads to the formation of long-lived mobile charge carriers with characteristic transient absorption signatures similar to those that have been observed in P3HT:PCBM bulk heterojunction blends. While Ï -stacking interactions between neighboring P3HT chains are weak in the micelles, they are strong in thin films drop-cast from ortho-dichlorobenzene. Here, PCB-P3HT self-assembles into a network of long fibers, clearly seen in atomic force microscopy images. Ultrafast charge separation occurs also for the fibrous morphology, but the transient absorption experiments show fast loss of part of the charge carriers due to intensity-induced recombination and annihilation processes and monomolecular interfacial trap-mediated or geminate recombination. The yield of the long-lived charge carriers in the highly organized fibers is however comparable to that obtained with annealed P3HT:PCBM blends. PCB-P3HT can therefore be considered as an active material in organic photovoltaic devices.
Even flow: Photoinduced symmetry-breaking charge separation takes place in a few picoseconds in a 1,3-bis(perylene)propane dyad in polar solvents. Polarized transient absorption measurements show that the direction of the charge flow is random and entirely governed by the fluctuations of the solvent orientation around the dyad.
Several novel aromatic ketone-based two-photon initiators containing triple bonds and dialkylamino groups were synthesized and the structure-activity relationships were evaluated. Branched alkyl chains were used at the terminal donor groups to improve the solubility in the multifunctional monomers. Because of the long conjugation length and good coplanarity, the evaluated initiators showed large two-photon cross section values, while their fluorescence lifetimes and quantum yields strongly depend on the solvent polarity. All novel initiators exhibited high activity in terms of two-photon-induced microfabrication. This is especially true for fluorenone-based derivatives, which displayed much broader processing windows than well-known highly active initiators from the literature and commercially available initiators. While the new photoinitiators gave high reactivity in two-photon-induced photopolymerization at concentration as low as 0.1% wt, these compounds are surprisingly stable under one photon condition and nearly no photo initiation activity was found in classical photo DSC experiment.
Ba2.2Ca0.8Mg4F14, a new “solid solution stabilized” matrix for an intense blue phosphor
F. Kubel, M. Pantazi and H. Hagemann Crystal Research and Technology, 46 (9) (2011), p899-905 Keywords: solid solution stabilized compound, fluoride, blue phosphor
DOI:10.1002/crat.201000624 | unige:16784 | Article PDF
Barium calcium magnesium fluoride (Ba2(BaxCa1-x)Mg4F14, x=0.19-0.26) has been synthesized at 850 °C from precursors prepared by the solution precipitation method. Single crystals with composition of Ba2.200(2)Ca0.800(2)Mg4F14were obtained after prolonged heating. Lattice parameters from single crystal data are a = 12.4203(8) and c = 7.4365(5) à [tetragonal, space group P42/mnm (No. 136)]. They increase with increasing barium concentration within a given stability window. The structure is built of a network of MgF6 octahedra forming a pyrochlore related channel system and isolated fluorine ions. Within the channels, heavy alkaline earth ions are located. The wide channel is filled with off-center positioned barium ions. The channel with a narrow cross section hosts both ions, Ca2+and Ba2+. The structure is isotypic with Pb3Nb4O12F2 but has a different coordination around Ba/Ca and Pb, respectively. Doped with â¼1% Eu(II), the compound shows intense blue luminescence under UV activation.
Several assertions which are incorrect or might be misleadingly interpreted as well as omissions of issues concerning the non-additive kinetic energy potential made by Fux et al. are analyzed. They concern issues of great importance for any computational method based on the orbital-free embedding theory: evaluation of the total energy, approximating the non-additive kinetic potential, exact properties of non-additive kinetic energy potential. In a nutshell, the authors do not distinguish between two different quantities: the functional, i.e., the correspondence assigning the non-additive kinetic potential to a pair of electron densities and the function (the potential itself).
The p53 tumour suppressor gene, the most frequently mutated gene in human cancer, encodes a transcription factor that contains sequence-specific DNA binding and homo-tetramerization domains. Interestingly, the affinities of p53 for specific and non-specific DNA sites differ by only one order of magnitude, making it hard to understand how this protein recognizes its specific DNA targets in vivo. We describe here the structure of a p53 polypeptide containing both the DNA binding and oligomerization domains in complex with DNA. The structure reveals that sequence- specific DNA binding proceeds via an induced fit mechan- ism that involves a conformational switch in loop L1 of the p53 DNA binding domain. Analysis of loop L1 mutants demonstrated that the conformational switch allows DNA binding off-rates to be regulated independently of affinities. These results may explain the universal prevalence of conformational switching in sequence-specific DNA binding proteins and suggest that proteins like p53 rely more on differences in binding off-rates, than on differences in affinities, to recognize their specific DNA sites.
Photoinduced electron transfer reactions: from the elucidation of old problems towards the exploration of interfaces
M. Fedoseeva, J. Grilj, O. Kel, M. Koch, R. Letrun, V. Markovic, I. Petkova, S. Richert, A. Rosspeintner, P. Sherin, D. Villamaina, B. Lang and E. Vauthey Chimia, 65 (2011), p350-352
DOI:10.2533/chimia.2011.350 | unige:16760 | Abstract | Article PDF
The activities of our research group in the field of photoinduced electron transfer reactions are discussed and illustrated by several examples
Self-Assembled Plasmonic Core-Shell Clusters with an Isotropic Magnetic Dipole Response in the Visible Range
S. Mühlig, A. Cunningham, S. Scheeler, C. Pacholski, T. Bürgi, C. Rockstuhl and F. Lederer ACS Nano, 5 (8) (2011), p6586-6592
DOI:10.1021/nn201969h | unige:16819 | Abstract | Article HTML | Article PDF
We theoretically analyze, fabricate, and characterize a three-dimensional plasmonic nanostructure that exhibits a strong and isotropic magnetic response in the visible spectral domain. Using two different bottom-up approaches that rely on self-organization and colloidal nanochemistry, we fabricate clusters consisting of dielectric core spheres, which are smaller than the wavelength of the incident radiation and are decorated by a large number of metallic nanospheres. Hence, despite having a complicated inner geometry, such a coreâshell particle is sufficiently small to be perceived as an individual object in the far field. The optical properties of such complex plasmonic coreâshell particles are discussed for two different core diameters.
A solid solution of magnesium and manganese borohydrides was studied by in situ synchrotron radiation X-ray powder diffraction and infrared spectroscopy. A combination of thermogravimetry, mass and infrared spectroscopy, and atomic emission spectroscopy were applied to clarify the thermal gas desorption of pure Mn(BH4)2 and a solid solution of composition Mg0.5Mn0.5(BH4)2. MgxMn(1âx)(BH4)2 (x = 0â0.8) conserves the trigonal structure of Mn(BH4)2 at room temperature. Manganese is dissolved in the hexagonal structure of α-Mg(BH4)2, with the upper solubility limit not exceeding 10 mol.% at room temperature. There exists a two-phase region of trigonal and hexagonal borohydrides within the compositional rangex = 0.8â0.9 at room temperature. Infrared spectra show splitting of various vibrational modes, indicating the presence of two cations in the trigonal MgxMn(1âx)(BH4)2 solid solutions, as well as the appearance of a second phase, hexagonal α-Mg(BH4)2, at higher magnesium contents. All vibrational frequencies are shifted to higher values with increasing magnesium content. The decomposition temperature of the trigonal MgxMn(1âx)(BH4)2 (x = 0â0.8) does not vary significantly as a function of the magnesium content (433â453 K). The desorbed gas contains mostly hydrogen and 3â7.5 mol.% diborane B2H6, as determined from analyses of the Mn(BH4)2 and Mg0.5Mn0.5(BH4)2 samples. An eutectic relation between α-Mg(BH4)2 and LiBH4 is observed. The solid solution MgxMn(1âx)(BH4)2 is a promising material for hydrogen storage as it decomposes at a similar temperature to Mn(BH4)2, i.e. at a much lower temperature than pure Mg(BH4)2 without significantly losing hydrogen weight capacity thanks to substitution of Mn by Mg up to 80 mol.%. The questions of diborane release and reversibility remain to be addressed.
«La chimie, ça reste une science expérimentale!» Entretien avec le professeur Jacques Weber
J. Weber Chimia, 65 (5) (2011), p362-365
DOI:10.2533/chimia.2011.362
The synthesis of a novel alkali-metal aluminium borohydride NaAl(BH4)xCl4âx from NaBH4 and AlCl3 using a solid state metathesis reaction is described. Structure determination was carried out using synchrotron powder diffraction data and vibrational spectroscopy. An orthorhombic structure (space group Pmn21) is formed which contains Na+ cations and complex [Al(BH4,Cl)4]âanions. Due to the high chlorine content (1 â¤Â x ⤠1.43) the hydrogen density of the borohydride is only between 2.3 and 3.5 wt.% H2 in contrast to the expected 14.6 wt.% for chlorine free NaAl(BH4)4. The decomposition of NaAl(BH4)xCl4âx is observed in the target range for desorption at about 90 °C by differential scanning calorimetry (DSC), in situ Raman spectroscopy and synchrotron powder X-ray diffraction. Thermogravimetric analysis (TG) shows extensive mass loss indicating the loss of H2 and B2H6 at about 90 °C followed by extensive weight loss in the form of chloride evaporation.
YMn2 forms either interstitial YMn2Hx hydrides for x ⤠4.5 or a complex YMn2H6 hydride when submitted to high hydrogen pressure. These compounds have been studied by inelastic neutron scattering (INS) in order to clarify the different modes of H vibration. The INS spectra of YMn2Hx hydrides are strongly dependent on the H content. YMn2H6 and YMn2D6 show broad bands, also observed by Raman and IR spectroscopy, assigned to HâMnâH (or D) and MnâH bending and stretching modes. Both ErMn2D6 and ErMn1.8Fe0.2D6 show, in addition to the H vibration mode, an intense band at 215 cmâ1 which has been attributed to a magnetic excitation of Er3+ in view of its momentum transfer dependence.
Size Exclusion Chromatography (SEC) on a semi-preparative scale (10 mg and more) was used to size-select ultrasmall gold nanoclusters (< 2 nm) from polydisperse mixtures. In particular, the ubiquitous byproducts of the etching process towards Au38(SR)24 (SR: thiolate) clusters were separated and gained in high monodispersity (based on mass spectrometry). The isolated fractions were characterized by UV/Vis spectroscopy, MALDI mass spectrometry and electron microscopy. Most notably, the separation of Au38(SR)24 and Au40(SR)24 clusters is demonstrated.
Nanofluidshave been proposed to improve the performance of microchannel heatsinks. In this paper, we present a systematic characterization ofaqueous silica nanoparticle suspensions with concentrations up to 31Â Â vol %. Wedetermined the particle morphology by transmission electron microscope imaging andits dispersion status by dynamic light scattering measurements. The thermophysicalproperties of the fluids, namely, their specific heat, density, thermalconductivity, and dynamic viscosity were experimentally measured. We fabricated microchannelheat sinks with three different channel widths and characterized theirthermal performance as a function of volumetric flow rate forsilica nanofluids at concentrations by volume of 0%, 5%, 16%,and 31%. The Nusselt number was extracted from the experimentalresults and compared with the theoretical predictions considering the changeof fluids bulk properties. We demonstrated a deviation of lessthan 10% between the experiments and the predictions. Hence, standardcorrelations can be used to estimate the convective heat transferof nanofluids. In addition, we applied a one-dimensional model ofthe heat sink, validated by the experiments. We predicted thepotential of nanofluids to increase the performance of microchannel heatsinks. To this end, we varied the individual thermophysical propertiesof the coolant and studied their impact on the heatsink performance. We demonstrated that the relative thermal conductivity enhancementmust be larger than the relative viscosity increase in orderto gain a sizeable performance benefit. Furthermore, we showed thatit would be preferable to increase the volumetric heat capacityof the fluid instead of increasing its thermal conductivity.
For nine solvents of various polarity (from cyclohexane to water), the solvatochromic shifts of the lowest absorption band of coumarin 153 are evaluated using a computational method based on frozen-density embedding theory [Wesolowski and Warshel, J. Chem Phys., 1993, 97, 9050, and subsequent articles]. In the calculations, the average electron density of the solvent ãÏB(râ)ã is used as the frozen density. ãÏB(râ)ã is evaluated using the statistical-mechanical approach introduced in Kaminski et al., J. Phys. Chem. A, 2010, 114, 6082. The small deviations between experimental and calculated solvatochromic shifts (the average deviation equals to about 0.02 eV), confirm the adequacy of the key approximations applied: (a) in the evaluation of the average effect of the solvent on the excitation energy, using the average density of the solvent instead of averaging the shifts over statistical ensemble and (b) using the approximant for the bi-functional of the non-electrostatic component of the orbital-free embedding potential, are adequate for chromophores which interact with the environment by non-covalent bonds. The qualitative analyses of the origin of the solvatochromic shifts are made using the graphical representation of the orbital-free embedding potential.
Shifts in the excitation energy of the organic chromophore, cis-7-hydroxyquinoline (cis-7HQ), corresponding to the ÏâÏ* transition in cis-7HQ and induced by the complexation with a variety of small hydrogen-bonded molecules, obtained with the frozen-density embedding theory (FDET), are compared with the results of the supermolecular equation-of-motion coupled-cluster (EOMCC) calculations with singles, doubles, and non-iterative triples, which provide the reference theoretical data, the supermolecular time-dependent density functional theory (TDDFT) calculations, and experiment. Unlike in the supermolecular EOMCC and TDDFT cases, where each complexation-induced spectral shift is evaluated by performing two separate calculations, one for the complex and another one for the isolated chromophore, the FDET shifts are evaluated as the differences of the excitation energies determined for the same many-electron system, representing the chromophore fragment with two different effective potentials. By considering eight complexes of cis-7HQ with up to three small hydrogen-bonded molecules, it is shown that the spectral shifts resulting from the FDET calculations employing non-relaxed environment densities and their EOMCC reference counterparts are in excellent agreement with one another, whereas the analogous shifts obtained with the supermolecular TDDFT method do not agree with the EOMCC reference data. The average absolute deviation between the complexation-induced shifts, which can be as large, in absolute value, as about 2000 cm-1, obtained using the non-relaxed FDET and supermolecular EOMCC approaches that represent two entirely different computational strategies, is only about 100 cm-1, i.e., on the same order as the accuracy of the EOMCC calculations. This should be contrasted with the supermolecular TDDFT calculations, which produce the excitation energy shifts that differ from those resulting from the reference EOMCC calculations by about 700 cm-1 on average. Among the discussed issues are the choice of the electronic density defining the environment with which the chromophore interacts, which is one of the key components of FDET considerations, the basis set dependence of the FDET, supermolecular TDDFT, and EOMCC results, the usefulness of the monomer vs supermolecular basis expansions in FDET considerations, and the role of approximations that are used to define the exchange-correlation potentials in FDET and supermolecular TDDFT calculations.
The femtosecond-resolved evolution of the emission spectrum of the important conjugated polymer poly(3-hexylthiophene) (P3HT) is presented. Detailed fluorescence up-conversion spectroscopy was performed on P3HT solid-state films and on P3HT in chlorobenzene solution. Two excitation wavelengths and several emission wavelengths, covering the entire fluorescence spectrum, were used. The data were complemented by polarization-sensitive measurements. Our global analysis allowed a reconstruction of the time-resolved emission spectra with 200 fs temporal resolution, so that spectral changes due to the early relaxation processes following ÏâÏ* interband absorption in the pristine polymer could be comprehensively characterized. Absorption occurs in isolated polymer chains in solution and in the solid state (including interchain interactions) for the film. In both cases, we find evidence of delocalization of the electrons and holes formed in the energy bands directly after photoexcitation with excess energy. This is followed by ultrafast (~100 fs) self-localization of the primary photoexcitation and by relatively slow exciton formation (~1 ps). Further relaxation occurs with time constants ranging from hundreds of femtoseconds to tens of picoseconds, due to exciton hopping to sites with lower energy and to a slow conformational planarization of the polymer backbone. Depolarization, a spectral red shift, and important changes in the vibronic structure are observed as a consequence of this relaxation. Finally, relaxed intrachain and interchain singlet excitons are formed in solution and film, respectively, on a 100â200 ps time scale. They decay with a ~500 ps time constant, by intersystem crossing in solution and by nonradiative recombination in the film. Our results are consistent with and strongly support the conclusions we obtained from a similar time-resolved fluorescence study of the polymer PCDTBT (J. Am. Chem. Soc.2010, 132, 17459): ultrafast charge separation in polymer:fullerene blends seems to occur before localization of the primary excitation to form a bound exciton.
The fluorescence lifetime of the radical cation of N,N,Nâ²,Nâ²-tetramethyl-p-phenylenediamine (Wurster's blue) decreases from 260â ps at 82â K to 200â fs at room temperature. Calculations indicate a small barrier between the excited-state minimum (D1 min) and a conical intersection (CI) of the excited and ground state potentials. The intersection is reached within 200â fs upon torsion of one of the CâN bonds.
A novel N-substituted 4-methoxy-1,8-naphthalimide (NAFTA 8) especially designed for fluorescent labeling of gold nanoparticles has been synthesized. NAFTA 8 bears a long methylene chain at the imide N atom and has a terminal SH group, which enables its chemical binding to gold nanostructures. The longest wavelength absorption maximum of NAFTA 8 in chloroform is at 370 nm, the fluorescent maximum is at 430 nm and the fluorescent quantum yield is 0.95. The newly synthesized fluorophore is applied for functionalization of gold nanoparticles with diameter 1.5 ± 0.5 nm prepared through chemical reduction. The obtained Monolayer Protected Clusters are characterized by elemental analysis, TEM, XPS, FT-IR, absorption and fluorescence spectroscopy. The performed investigations provide evidence for the formation of chemical bond between the thiol ligand and the gold surface. They also show that the obtained metal/dielectric 3D structures are highly fluorescent.
Using bottom-up and self-assembly processes, large scale layered arrays of strongly coupled gold nanoparticles with controllable dimensions were fabricated. By carefully adjusting the distance between adjacent gold nanoparticle arrays, it is possible to control the coupling of the localized surface plasmon polariton resonance as sustained by individual gold nanoparticles. A greater interaction is observed at smaller separations, leading to a well pronounced shift in the spectral position of resonances that can be adjusted with high precision. Simulations showed good agreement with experimental observations in an in-depth investigation of such structures, suggesting minimal separations of only one nanometer are achieved.
The excited-state dynamics of eosin B (EB) at dodecane/water and decanol/water interfaces has been investigated with polarization-dependent and time-resolved surface second harmonic generation. The results of the polarization-dependent measurements vary substantially with (1) the EB concentration, (2) the age of the sample, and (3) the nature of the organic phase. All of these effects are ascribed to the formation of EB aggregates at the interface. Aggregation also manifests itself in the time-resolved measurements as a substantial shortening of the excited-state lifetime of EB. However, independently of the dye concentration used, the excited-state lifetime of EB at both dodecane/water and decanol/water interfaces is much longer than in bulk water, where the excited-state population undergoes hydrogen-bond-assisted non-radiative deactivation in a few picoseconds. These results indicate that hydrogen bonding between EB and water molecules at liquid/water interfaces is either much less efficient than in bulk water or does not enhance non-radiative deactivation. This strong increase of the excited-state lifetime of EB at liquid/water interfaces opens promising avenues of applying this molecule as a fluorescent interfacial probe.
Near-Infrared to Visible Light Upconversion in a Trinuclear d-f-d Complex
L. Aboshyan-Sorgho, C. Besnard, P. Pattison, K.R. Kittilstved, A. Aebischer, J.-C.G. Bünzli, A. Hauser and C. Piguet Angewandte Chemie International Edition, 50 (2011), p4108-4112 Keywords: helical structures;lanthanides;photochemistry;self-assembly;upconversion
DOI:10.1002/anie.201100095 | unige:15714 | Abstract | Article PDF
The connection of two CrIII sensitizers around a central ErIII acceptor in a self-assembled cation provides high local metal concentrations that favor efficient nonlinear energy transfer upconversion luminescence (see picture). Upon selective low-energy near-infrared irradiation of CrIII-centered transitions, 1 displays an unprecedented molecular two-photon upconverted green ErIII-centered emission.
We investigated by optical microscopy the thermal spin transition in single crystals of [Fe(bbtr)3](ClO4)2 (bbtr = 1,4-di(1,2,3-triazol-1-yl) butane). The growth of the low-spin phase was observed for different crystal orientations and sizes. The process always started from a corner of the crystal but its further development depended on the size, shape and thermal history of the crystal. In crystals of smaller size, under isothermal conditions, the low-spin phase developed in a continuous way, through the propagation of a rather well defined transformation front, with a higher propagation velocity inside the planes perpendicular to the c axis. In larger crystals the spontaneous occurrence of inhomogeneous stresses led to a stepwise propagation process.
The photocatalytic degradation of l-asparagine and l-glutamic acid over Au/TiO2 and TiO2 catalysts was investigated in situ by attenuated total reflection infrared (ATR-IR) in combination with modulation excitation spectroscopy. Oxalate was detected on the catalyst surface, which has not been reported before for degradation of amino acids by studies focusing on intermediates in solution. The ATR-IR spectra provide valuable information on the fate of the nitrogen. Ammonium was detected, in agreement with previous studies. Most importantly, strong signals of cyanide were observed, and this assignment has been corroborated by 15N labeling experiments. Cyanide was not reported before, to the best of our knowledge, for the photocatalytic degradation of amino acids. Cyanide was formed in the presence and the absence of gold particles on the TiO2 surface. The cyanide leads to leaching of gold via Au(CN)2â species that were detected in solution by mass spectrometry.
Shining Light at Working Interfaces and Chiral Nanoparticles
T. Bürgi Chimia, 65 (3) (2011), p157-167 Keywords: attenuated total reflection;chiral surfaces;nanoparticles;photocatalysis;spectroscopy
DOI:10.2533/chimia.2011.157 | unige:15782 | Abstract | Article PDF
In this article we present an overview of our recent research in the fields of in situ spectroscopy, nanomaterials and chirality. Our research focuses around the spectroscopic investigation of chemical reactions taking place at solid-liquid interfaces. This research goesh and in hand with the development of experimental techniques that enable us to study interface phenomena in situ. Using such techniques we try to shed light on photocatalytic reactions like the decomposition of organic pollutants in water or the reduction of carbon dioxide. We are moreover interested in chiral surfaces and their ability to discriminate betweenen antiomers. Again this relies on special techniques that highlight the enantiodiscriminating surface-adsorbate interactions. We further more seek to transfer chirality from adsorbates to metal nanoparticles. The latter are probed by chiroptical techniques, particularly also vibrational circular dichroism (VCD). Finally, we aim at preparing metamaterials with tailored optical properties by organizing plasmonic particles in two and three dimensions.
The photophysics of two dyes from the xanthene family, eosin B (EB), and eosin Y (EY) has been investigated in various solvents by femtosecond transient absorption spectrosco- py, first, to clarify the huge disparity of the EB fluorescence lifetimes reported in literature, and, second, to understand the mechanism responsible for the ultrafast excited-state deactivation of EB in water. The excited-state lifetime of EB was found to be much shorter in water and in other protic solvents, due to the occurrence of hydrogen-bond assisted nonradiative deactivation. This mechanism is associated with the hydrogen bonds between the solvent molecules and the nitro groups of EB, which become stronger upon optical excitation due to the charge-transfer character of the excited-state. This process is not operative with EY, where the nitro groups are replaced by bromine atoms. Therefore, the excited-state lifetime of EB in solution is directly related to the strength of the solvent as a hydrogen-bond donor, offering the possibility to build a corresponding scale based on the fluorescence quantum yield or lifetime of EB. This scale of hydrogen-bonding strength could be especially useful for studies of liquid interfaces by time-resolved surface second harmonic generation.
The synthesis and the photophysical properties of the complex [Ru(TTF-dppz)2(Aqphen)]2+(TTF = tetrathiafulvalene, dppz = dipyrido-[3,2-a:2â²,3â²-c]phenazine, Aqphen = anthraquinone fused to phenanthroline via a pyrazine bridge) are described. In this molecular triad excitation into the metalâligand charge transfer bands results in the creation of a long-lived charge separated state with TTF acting as electron donor and anthraquinone as terminal acceptor. The lifetime of the charge-separated state is 400 ns in dichloromethane at room temperature. A mechanism for the charge separation involving an intermediate charge-separated state is proposed based on transient absorption spectroscopy.
Ground and Excited State Double Hydrogen Transfer in Symmetric and Asymmetric Potentials: Comparison of 2,7,12,17-Tetra-n-propylporphycene with 9-Acetoxy-2,7,12,17-tetra-n-propylporphycene
P. Fita, P. Garbacz, M. Nejbauer, C. Radzewicz and J. Waluk Chemistry - A European Journal, 17 (13) (2011), p3672-3678 Keywords: density functional theory;hydrogen transfer;porphycenes;tautomerism;time-resolved anisotropy
DOI:10.1002/chem.201002931 | unige:14939 | Abstract | Article PDF
Analysis of time-resolved anisotropy of transient absorption enabled determination of room temperature ground and excited state rate constants for intramolecular double hydrogen transfer in two similar porphycenes, one of them with symmetric and the other, with asymmetric character of a double minimum potential for hydrogen motion. The perturbation preserves a quasi-symmetric minimum in S0, but the rate decreases approximately two times. In S1, the perturbed potential becomes strongly asymmetric, and the downhill hydrogen transfer occurs with a rate higher than that observed for a symmetrical compound.
The excited-state dynamics of aminostilbazolium dyes is known to be dominated by nonradiative deactivation through large-amplitude motion. In order to identify the coordinate(s) responsible for this process, the excited-state lifetimes of two dialkylaminostyryl-methylpyridinium iodides have been measured at liquidâliquid interfaces using time-resolved surface second harmonic generation. We found that the decay time of the excited-states of both compounds was increasing with the viscosity of the apolar phase, consisting of n-alkanes of varying length, but was unaffected by that of the polar phase, made of water/glycerol mixtures. This indicates that the nonradiative deactivation is associated with the twist of the dialkylaniline group, which is located in the apolar part of the molecule.
Whereas the neat polymeric iron(II) compound [Fe(bbtr)3](ClO4)2, bbtr = 1,4-di(1,2,3-triazol-1-yl)butane, shows a quantitative spin transition triggered by a crystallographic phase transition centered at 107 K with a 13 K wide hysteresis, the iron(II) complexes in the diluted mixed crystals [FexZn1âx(bbtr)3](ClO4)2, x = 0.02 and 0.1, stay predominantly in the 5T2 high-spin state down to cryogenic temperatures. However, the 1A1 low-spin state can be populated as metastable state via irradiation into the spin-allowed 5T2â5E ligand-field transition of the high-spin species in the near-infrared. The quantum efficiency of the light-induced conversion is approximately 10% at low temperatures and decreases rapidly above 160 K. The lifetime of the light-induced low-spin state decreases from 15 days at 40 K to 30 ns at 220 K, that is, by 14 orders of magnitude. In the high-temperature regime the activation energy for the low-spinâhigh-spin relaxation is 1840(20) cmâ1.
The structural and vibrational properties of the isostructural compounds Ca2FeH6Â and Sr2RuH6Â are determined by periodic DFT calculations and compared with their previously published experimental crystal structures as well as new experimental vibrational data. The analysis of the vibrational data is extended to the whole series of alkaline-earth iron and ruthenium hydrides A2TH6Â (A = Mg,Ca,Sr; T = Fe, Ru) in order to identify correlations between selected frequencies and the T-H bond length. The bulk moduli of Ca2FeH6Â and Sr2RuH6Â have also been determined within DFT. Their calculated values prove to compare well with the experimental values reported for Mg2FeH6Â and several other compounds of this structure.
Experimental evidence of librational vibrations determining the stability of calcium borohydride
A. Borgschulte, R. Gremaud, A. Züttel, P. Martelli, A. Remhof, A.J. Ramirez-Cuesta, K. Refson, E.G. Bardaji, W. Lohstroh, M. Fichtner, H. Hagemann and M. Ernst Physical Review B, 83 (2) (2011), p24102
DOI:10.1103/PhysRevB.83.024102 | unige:14989 | Abstract | Article PDF
The high energy of hydrogen vibrations in solids is the origin of their strong impact on thermodynamic properties. The peculiar structure of complex hydrides amplifies this impact. We shed light on the vibrational properties of three allotropes of Ca(BH4)2 using density-functional theory calculations, infrared spectroscopy, and inelastic neutron scattering. We show that the vibrational properties of Ca(BH4)2 depend on the specific phase and are hitherto the origin of their differences in stability.
We propose a new approach to determine a suitable zeroth-order wavefunction for multiconfigurational perturbation theory. The same ansatz as in complete active space (CAS) wavefunction optimization is used but it is split in two parts, a principal space (A) and a much larger extended space (B). Löwdin's partitioning technique is employed to map the initial eigenvalue problem to a dimensionality equal to that of (A) only. Combined with a simplified expression for the (B) portion of the wavefunction, we are able to drastically reduce the storage and computational demands of the wavefunction optimization. This scheme is used to produce reference wavefunctions and energies for subsequent second-order perturbation theory (PT2) corrections. Releasing the constraint of computing the exact CAS energy and wavefunction prior to the PT2 treatment introduces a nonstandard paradigm for multiconfigurational methods. Based on the results of test calculations, we argue that principal parts with only few percents of the total number of CAS configurations could provide final multiconfigurational PT2 energies of the same accuracy as in the standard paradigm. In the future, algorithmic improvements for this scheme will bring into reach active spaces much beyond the present limit of CAS-based methods, therefore allowing for accurate studies of systems featuring strong correlation.
New fundamental experimental studies on α-Mg(BH4)2 and other borohydrides
H. Hagemann, V. D'Anna, J.-P. Rapin, R. Cerný, Y. Filinchuk, K.C. Kim, D.S. Sholl and S.F. Parker Journal of Alloys and Compounds, 509 (2) (2011) Keywords: hydrogen storage materials; vibrational spectroscopy; synchrotron X-ray diffraction
DOI:10.1016/j.jallcom.2010.10.068 | unige:16970 | Article HTML | Article PDF
Several new studies of Mg(BH4)2 are reported. A 1:1 LiBH4:Mg(BH4)2 mixture was studied by in situ synchrotron X-ray diffraction and reveals an eutectic behavior with the eutectic composition more rich in Mg(BH4)2, and the eutectic temperature lower than 456Â K. No dual cation compound was observed in this experiment.
New vibrational spectra including INS data have been obtained and are compared with theoretical DFT calculations and recent NMR studies, showing good agreement.
Polarized Raman and Hyperpolarizability studies of Hydroxyethylammonium (L) tartrate monohydrate for quadratic nonlinear optics
R. Nagalakshmi, V. Krishnakumar, H. Hagemann and S. Muthunatesan Journal of Molecular Structure, 988 (2011), p17-23 Keywords: solution growth; X ray diffraction; vibrational spectroscopy; polarized Raman, hyperpolarizability
DOI:10.1016/j.molstruc.2010.11.056 | unige:14817 | Abstract | Article PDF
Single crystals of Hydroxyethylammonium L â tartrate monohydrate [HEALT] have been grown by slow evaporation technique using water as a solvent. The structural and vibrational properties of the crystals were studied. Besides these characterizations ab initio quantum chemical calculations have been performed at HF/6-31G (d) level to derive first order hyperpolarizability. It is shown that the first order hyperpolarizability is found to be 14.2 times more than that of urea. The characteristic vibrational frequencies obtained from polarized Raman spectra in different scattering configurations have been assigned based on the complete factor group analysis. Vibrational analysis of IR and Raman reveals that the charge transfer interaction must be responsible for nonlinear optical (NLO) properties of the present system. The UV absorption measurements have also been carried out to confirm the utility of the material for optical applications.
The importance of the nonelectrostatic component of the embedding potential is investigated by comparing the complexation induced shifts of the iso-g obtained in embedding calculations to its supermolecular counterparts. The analyses are made in view of such multilevel simulations, for which supermolecular strategy is either impractical or impossible, such as the planned simulations for the whole enzyme ferredoxin oxidoreductase. For the biliverdin radical surrounded by a few amino acids, it is shown that the embedding potential comprising only Coulomb terms fails to reproduce even qualitatively the shifts evaluated from supermolecular calculations. The nonelectrostatic component of the exact embedding potential is a bifunctional of two electron densities [Wesolowski and Warshel, J. Phys. Chem.1993, 97, 8050; Wesolowski, Phys. Rev. A2008, 77, 012504]. Therefore we analyze in detail both the quality of the approximant for the bifunctional and the importance of the choice of the electron densities at which it is evaluated in practical calculations.
The recently developed second-order perturbation theory restricted active space (RASPT2) method has been benchmarked versus the well-established complete active space (CASPT2) approach. Vertical excitation energies for valence and Rydberg excited states of different groups of organic (polyenes, acenes, heterocycles, azabenzenes, nucleobases, and free base porphin) and inorganic (nickel atom and copper tetrachloride dianion) molecules have been computed at the RASPT2 and multistate (MS) RASPT2 levels using different reference spaces and compared with CASPT2, CCSD, and experimental data in order to set the accuracy of the approach, which extends the applicability of multiconfigurational perturbation theory to much larger and complex systems than previously. Relevant aspects in multiconfigurational excited state quantum chemistry such as the valenceâRydberg mixing problem in organic molecules or the double d-shell effect for first-row transition metals have also been addressed.
The photophysics and excited-state dynamics of two dyads consisting of either a free-base or a zinc-tetraphenylporphyrin linked through a rigid bridge to a core-substituted naphthalenediimide (NDI) have been investigated by femtosecond-resolved spectroscopy. The absorption and fluorescence spectra differ substantially from those of the individual units, pointing to a substantial coupling and to a delocalisation of the excitation over the whole molecule, as confirmed by quantum chemistry calculations. A strong dependence of their excited-state dynamics on the solvent polarity has been observed. In toluene, the fluorescence quantum yield of the dyads is of the order of a few percent and the main decay channel of the emitting state is proposed as intersystem-crossing to the triplet state. However, in a medium polarity solvent like dichloromethane, the emitting state undergoes charge separation from the porphyrin to the NDI unit within 1â3 ps, and the ensuing charge-separated state recombines in about 10â20 ps. This solvent dependence can be explained by the weak driving force for charge separation in polar solvents and the large electronic coupling between the porphyrin and NDI moieties, making charge separation a solvent-controlled adiabatic process.
Identification of New Aromatic Compounds in the New Zealand Manuka Honey by Gas Chromatography-Mass Spectrometry
S. Daher and F.O. Gülaçar E-Journal of Chemistry, 7 (S1) (2010), p7-14 Keywords: Manuka honey; aromatic compounds; solid phase microextraction; GC-CMS
DOI:10.1155/2010/472769 | unige:21623 | Abstract | Article PDF
Analysis of aromatic compounds in the New Zealand manuka honey was carried out by solid phase microextraction followed by gas chromatography-mass spectrometry. A total of 38 compounds were detected. Seven of them such as; 1,4-bis(x-methoxyphenyl)-but-2-en-1-one, 1,5-bis(x-methoxyphenyl)-pent-3-en-1-one, 1,4-bis(x-methoxyphenyl)-1-pentanone, 1,6-bis(x-methoxyphenyl)-3-heptene, 1,6-bis(x-methoxyphenyl)-hex-2(3 or 4)-en-1-one and 2(3, 4 or 5)-hydroxy-1,6-bis(x-methoxyphenyl)-1-hexanone, had never before been identified as natural products. Their structures were deduced from the mass spectral data. Seven other compounds; 2,3-dimethoxynaphthalene, 4-(x-methoxyphenyl)-1-phenyl-1-butanone, desoxyanisoin, 2,6-dimethoxybenzoic acid benzyl ester, 4,4'-dimethoxystilbene, 3,3,4,5,5,8-hexamethyl-2,3,5,6-tetrahydro-s-indacene-1,7-dione and 1,5-bis(4-methoxyphenyl)-pentane-1,5-dione, were found in honey for the first time. Methyl syringate, ortho-methoxyacetophenone and 3-phenyllactic acid were the most abundant components.
Kinetic Control in the Chiral Recognition of Three-Bladed Propellers
C. Bonnot, E. Aubert, N. Banerji, J. Lacour, E. Espinosa and J.-C. Chambron Chemistry - A European Journal, 16 (19) (2010), p5706-5711 Keywords: cryptands;helical structures;ion pairs;kinetics;solvent effects
DOI:10.1002/chem.200903058 | unige:6579 | Abstract | Article PDF
The ion pair of the stereolabile C3-symmetric, i+o proton complex [1â H]+ of diaza-macropentacycle 1 and the configurationally stable Î-TRISPHAT ([Î-3]â) anion exists in the form of two diastereomers, namely, [Î-(1â H)][Î-3] and [Î-(1â H)][Î-3], the ratio of which, in terms of diastereomeric excess (de) decreases in the order [D8]THF (28â%)>CD2Cl2 (22â%)>CDCl3 (20â%)>[D8]toluene (16â%)>C6D6 (7â%)>[D6]acetone (0â%) at thermodynamic equilibrium. Except in the case of [D6]acetone, the latter is reached after a period of time that increases from 1â h ([D8]THF) to 24â h (CDCl3). Moreover, the initial value of the de of [1â H][Î-3] in CDCl3, before the thermodynamic equilibrium is reached, depends on the solvent in which the sample has been previously equilibrated (sample âhistoryâ). This property has been used to show that the crystals of [1â H][Î-3] formed by slow evaporation of CH2Cl2/CH3OH mixtures had 100â% de, which indicates that [1â H][Î-3] has enjoyed a crystallization-induced asymmetric transformation. Structural studies in solution (NMR spectroscopy) and in the gas phase by calculations at the semiempirical PM6 level of theory suggest that the optically active anion is docked on the i+ (endo) external side of the proton complex such that one of the aromatic rings of [Î-3]â is inserted into a groove of [1â H]+, a second aromatic ring being placed astride the outside i+ pocket. Solvent polarity controls the thermodynamics of inversion of the [1â H]+ propeller. However, both polarity and basicity control its kinetics. Therefore, the rate-limiting steps correspond to the ion-pair separation/recombination and [1â H]+/1deprotonation/protonation processes, rather than the inversion of [1â H]+, the latter being likely to take place in the deprotonated form (1).
Orbital-Free Embedding Effective Potential in Analytically Solvable Cases
A. Savin and T.A. Wesolowski
in "Progress in Theoretical Chemistry and Physics"
Advances in the Theory of Atomic and Molecular Systems, P. Piecuch, J. Maruani, G. Delgado-Barrio and S. Wilson, Springer,
19 (2010), p311-326 Keywords: embedding potential; density functional theory; kinetic energy functional; orbital-free embedding
DOI:10.1007/978-90-481-2596-8 | unige:14990
The effective embedding potential introduced by Wesolowski and Warshel [J. Phys. Chem., 97 (1993) 8050] depends on two electron densities: that of the environment (n B ) and that of the investigated embedded subsystem (n A ). In this work, we analyze this potential for pairs n A  and n B , for which it can be obtained analytically. The obtained potentials are used to illustrate the challenges in taking into account the Pauli exclusion principle.
Unidirectional Photoisomerization of Styrylpyridine for Switching the Magnetic Behavior of an Iron(II) Complex: A MLCT Pathway in Crystalline Solids
A. Tissot, M.-L. Boillot, S. Pillet, E. Codjovi, K. Boukheddaden and L.M. Lawson Daku Journal of Physical Chemistry C, 114 (49) (2010), p21715-21722
DOI:10.1021/jp106583f | unige:14853 | Abstract | Article HTML | Article PDF
The photoreactivity of two iron(II)âstyrylpyridine frameworks Fe(stpy)4(NCSe)2 (stpy = 4-styrylpyridine) has been investigated for the very first time in a crystalline solid. A quantitative cis-to-trans isomerization of stilbenoids is shown to occur in the confined environment of the inorganic solid. The photochromic reaction was driven by a visible excitation into the metal-to-ligand charge transfer absorption of the high-spin all-cis complex. The solid-state transformation is accompanied by a unit-cell volume increase and an amorphization. Interestingly, the photoproduct formed by irradiating the high-spin all-cis reactant undergoes a spin conversion when the temperature is decreased. This observation is related to the âligand-driven light-induced spin changeâ effect in a constrained environment.
Two tridentate and one bidentate binding strands have been anchored on a carbon atom to provide a new unsymmetrical tripodal ligand L for Ln(III) coordination. The ligand itself adopts a single conformation in solution stabilized by intramolecular hydrogen bonds evidenced in the solid state. The reaction of L with trivalent lanthanides provides different coordination complexes depending on the metal/ligand ratio. The speciation studies with selected lanthanides were performed in solution by means of NMR, ESMS, and spectrophotometric titrations. Differences in coordination properties along the lanthanide series were evidenced and may be associated with the changes in the ionic size. However, thermodynamic stability constants for the species of the same stoichiometry do not significantly vary. In addition, the structure of the dinuclear complex [Eu2L2]6+ has been elucidated in the solid state, where the complex crystallizes predominantly as an M-isomer. The crystal structure shows the coordination of two different ligands to each europium cation through tridentate strands, and the europium nine-coordinate sphere is completed with three solvent molecules. Finally, the results of photophysical investigations of [Eu2L2]6+ are in close agreement with the structural parameters determined by crystallography.
The nature and time evolution of the primary excitations in the pristine conjugated polymer, PCDTBT, are investigated by femtosecond-resolved fluorescence up-conversion spectroscopy. The extensive study includes data from PCDTBT thin film and from PCDTBT in chlorobenzene solution, compares the fluorescence dynamics for several excitation and emission wavelengths, and is complemented by polarization-sensitive measurements. The results are consistent with the photogeneration of mobile electrons and holes by interband Ï-Ï* transitions, which then self-localize within about 100 fs and evolve to a bound singlet exciton state in less than 1 ps. The excitons subsequently undergo successive migrations to lower energy localized states, which exist as a result of disorder. In parallel, there is also slow conformational relaxation of the polymer backbone. While the initial self-localization occurs faster than the time resolution of our experiment, the exciton formation, exciton migration, and conformational changes lead to a progressive relaxation of the inhomogeneously broadened emission spectrum with time constants ranging from about 500 fs to tens of picoseconds. The time scales found here for the relaxation processes in pristine PCDTBT are compared to the time scale (<0.2 ps) previously reported for photoinduced charge transfer in phase- separated PCDTBT:fullerene blends (Phys. Rev. B 2010, 81, 125210). We point out that exciton formation and migration in PCDTBT occur at times much longer than the ultrafast photoinduced electron transfer time in PCDTBT:fullerene blends. This disparity in time scales is not consistent with the commonly proposed idea that photoinduced charge separation occurs after diffusion of the polymer exciton to a fullerene interface. We therefore discuss alternative mechanisms that are consistent with ultrafast charge separation before localization of the primary excitation to form a bound exciton.
The photoinduced processes occurring after pulsed laser excitation of a series of donorâbridgeâacceptor molecules comprising a phenothiazine electron donor, variable-length fluorene bridges, and a rhenium(I) electron acceptor were investigated. A dyad with a single fluorene bridge unit exhibits electron transfer from phenothiazine to the rhenium(I) complex upon photoexcitation, whereas in dyads with fluorene oligomers bridge-localized triplet excited states are formed rather than electron transfer products. In the monofluorene-bridged system with a donorâacceptor distance of ca. 15 Ã , electron transfer occurs with a time constant of 1.9 ns. The equidistant electron transfer between the same donor and acceptor is considerably slower across a biphenyl bridge (3.9 ns) or a bi-p-xylene spacer (20 ns). This finding is interpreted in terms of different tunneling barrier heights associated with the charge transfer across the three different types of molecular bridges.
Structure and Characterization of KSc(BH4)4
R. Cerny, D.B. Ravnsbæk, G. Severa, Y. Filinchuk, V. D'Anna, H. Hagemann, D. Haase, J. Skibsted, C.M. Jensen and T.R. Jensen Journal of Physical Chemistry C, 114 (45) (2010), p19540-19549 Keywords: borohydrides; hydrogen storage; metathesis; X-ray diffraction
DOI:10.1021/jp106280v | unige:14680 | Abstract | Article HTML | Article PDF
A new potassium scandium borohydride, KSc(BH4)4, is presented and characterized by a combination of in situ synchrotron radiation powder X-ray diffraction, thermal analysis, and vibrational and NMR spectroscopy. The title compound, KSc(BH4)4, forms at ambient conditions in ball milled mixtures of potassium borohydride and ScCl3 together with a new ternary chloride K3ScCl6, which is also structurally characterized. This indicates that the formation of KSc(BH4)4 differs from a simple metathesis reaction, and the highest scandium borohydride yield (~31 mol %) can be obtained with a reactant ratio KBH4:ScCl3 of 2:1. KSc(BH4)4 crystallizes in the orthorhombic crystal system, a = 11.856(5), b = 7.800(3), c = 10.126(6) à , V = 936.4(8) à 3 at RT, with the space group symmetry Pnma. KSc(BH4)4 has a BaSO4 type structure where the BH4 tetrahedra take the oxygen positions. Regarding the packing of cations, K+, and complex anions, [Sc(BH4)4]â, the structure of KSc(BH4)4 can be seen as a distorted variant of orthorhombic neptunium, Np, metal. Thermal expansion of KSc(BH4)4 in the temperature range RT to 405 K is anisotropic, and the lattice parameter b shows strong nonlinearity upon approaching the melting temperature. The vibrational and NMR spectra are consistent with the structural model, and previous investigations of the related compounds ASc(BH4)4 with A = Li, Na. KSc(BH4)4 is stable from RT up to ~405 K, where the compound melts and then releases hydrogen in two rapid steps approximately at 460â500 K and 510â590 K. The hydrogen release involves the formation of KBH4, which reacts with K3ScCl6 and forms a solid solution, K(BH4)1âxClx. The ternary potassium scandium chloride K3ScCl6 observed in all samples has a monoclinic structure at room temperature, P21/a, a = 12.729(3), b = 7.367(2), c = 12.825(3) à , β = 109.22(2)°, V = 1135.6(4) à 3, which is isostructural to K3MoCl6. The monoclinic polymorph transforms to cubic at 635 K, a = 10.694 à (based on diffraction data measured at 769 K), which is isostructural to the high temperature phase of K3YCl6.
The thiolate-for-thiolate ligand exchange reaction between the stable Au38(2-PET)24 and Au40(2-PET)24 (2-PET: 2-phenylethanethiol) clusters and enantiopure BINAS (BINAS: 1,1â²-binaphthyl-2,2â²-dithiol) was investigated by circular dichroism (CD) spectroscopy in the UV/vis and MALDI mass spectrometry (MS). The ligand exchange reaction is incomplete, although a strong optical activity is induced to the resulting clusters. The clusters are found to be relatively stable, in contrast to similar reactions on [Au25(2-PET)18]â clusters. Maximum anisotropy factors of 6.6 à 10â4 are found after 150 h of reaction time. During the reaction, a varying ratio between Au38 and Au40 clusters is found, which significantly differs from the starting material. As compared to Au38, Au40 is more favorable to incorporate BINAS into its ligand shell. After 150 h of reaction time, an average of 1.5 and 4.5 BINAS ligands is found for Au38 and Au40 clusters, respectively. This corresponds to exchange of 3 and 9 monodentate 2-PET ligands. To show that the limited exchange with BINAS is due to the bidentate nature of the ligand, exchange with thiophenol was performed. The monodentate thiophenol exchange was found to be faster, and more ligands were exchanged when compared to BINAS.
The emission spectra of the solids [n-Bu4N]2Tc2X8 (X = Cl, Br) have been investigated at room temperature and 77 K. In each case, the emission originates in the 1δâδ* excited state, as with the rhenium homologues, but has a shorter lifetime.
We present the results of a quantum chemical and classical molecular dynamics simulation study of some solutions containing chloride salts of La3+, Gd3+, and Er3+ at various concentrations (from 0.05 to 5 M), with the purpose of understanding their structure and dynamics and analyzing how the coordination varies along the lanthanide series. In the LaâCl case, nine water molecules surround the central La3+ cation in the first solvation shell, and chloride is present only in the second shell for all solutions but the most concentrated one (5 M). In the Gd3+ case, the coordination number is ~8.6 for the two lowest concentrations (0.05 and 0.1 M), and then it decreases rapidly. In the Er3+ case, the coordination number is 7.4 for the two lowest concentrations (0.05 and 0.1 M), and then it decreases. The counterion Clâ is not present in the first solvation shell in the La3+ case for most of the solutions, but it becomes progressively closer to the central cation in the Gd3+ and Er3+ cases, even at low concentrations.
A combination of in situ synchrotron powder diffraction, energy minimization (DFT), and Raman and infrared spectroscopy confirmed porous interpenetrated 3D-framework structures of recently discovered alkali-metalâzinc borohydrides, AZn2(BH4)5 (A = Li, Na). In the less zinc rich NaZn(BH4)3 the 3D-framework structural model has been confirmed but with a slightly modified description giving an isolated triangular anion, [Zn(BH4)3]â, rather than a 1D anionic chain, {[Zn(BH4)3]n}nâ. Another polymorph of NaZn(BH4)3, isostructural to a new compound, LiZn(BH4)3, is proposed by energy minimization. Both compounds, the new NaZn(BH4)3 polymorph and LiZn(BH4)3, are, however, not observed experimentally at ambient pressure and in the temperature range of 100â400 K. The alkali-metalâzinc borohydride NaZn(BH4)3 containing the triangular anion [Zn(BH4)3]â is an equivalent of recently characterized alkali-metalâscandium borohydrides NaSc(BH4)4 and LiSc(BH4)4 based on the tetrahedral [Sc(BH4)4]â complex anion.
The crystal structure of recently reported Ba,F,Cl nanorods is shown to correspond to the structure of Ba7F12Cl2 (see picture), which can be prepared by several growth techniques.
Significant variation of the singlet-quintet intersystem crossing rate constant in an iron(II) high-spin complex as a function of temperature
I. Krivokapic, P. Chakraborty, R. Bronisz, C. Enachescu and A. Hauser Angewandte Chemie, 49 (45) (2010), p8509-8512
DOI:10.1002/anie.201004500 | unige:14713
In the dilute mixed-crystal system [Zn1âxFex(bbtr)3](ClO4)2, x=2 % (bbtr=1,4-di(1,2,3-triazol-1-yl)butane), the iron(II) centers are predominantly in the high-spin state. The low-spin state can be populated as a metastable state by irradiation with near-IR light; the rate constant of the low-spinâhigh-spin relaxation spans 14 orders of magnitude between 40 and 220 K
The ligand 2,4,6-tris(dimethoxyphosphonate)-1,3,5-triazine L has been synthesized and its single crystal X-ray structure determined. The occurrence of P=OÂ·Â·Â·Ï intermolecular interactions, suggested by the short P=O··· triazine distances of 3.16â3.35 à , is observed. The electrochemical reduction of the ligand shows its electron acceptor character by the formation of a stable radical anion. The hyperfine structure observed in the EPR spectra, combined with a theoretical DFT study, evidences the full delocalization of the unpaired electron mainly on the triazine core, with some participation of the phosphonate groups. Theoretical calculations are in agreement with the experimental values of the hyperfine coupling constants of 11.81 G for Aisoâ31P and 1.85 G for Aisoâ14N. Homopolymetallic complexes, formulated as {L[Cu(hfac)2]3} (1), 1â{L2[Co(hfac)2]3} (2) and 1â{L2[Mn(hfac)2]3} (3) (hfac = hexafluoroacetylacetonate), have been synthesized and structurally characterized.
The cis-[RuII(bpy)2(H2O)2]2+ Water-Oxidation Catalyst Revisited
X. Sala, M.Z. Ertem, L. Vigara, T.K. Todorova, W. Chen, R.C. Rocha, F. Aquilante, C.J. Cramer, L. Gagliardi and A. Llobet Angewandte Chemie International Edition, 49 (42) (2010), p7745-7747 Keywords: density functional calculations; energy conversion; reaction mechanisms; ruthenium; water oxidation
DOI:10.1002/anie.201002398 | unige:14721 | Abstract | Article PDF
The only operating mechanism in the oxidation of water to dioxygen catalyzed by the mononuclear cis-[RuII(bpy)2(H2O)2]2+ complex when treated with excess CeIV was unambiguously established. Theoretical calculations together with 18O-labeling experiments (see plot) revealed that it is the nucleophilic attack of water on a Ru=O group.
In this paper we use a recently proposed elastic model in order to study the competition between linear photoexcitation and cooperative relaxation in spin-crossover molecular magnets. The difference in molecular size between the two possible spin states, that is, the high-spin and the low-spin states, respectively, induces distortions of the crystal lattice. These determine the elastic interactions between molecules, treated here as connecting springs that are either compressed or extended from their equilibrium length, thus modulating the local probability for the high-spinâlow-spin relaxation. The crossover of individual molecules within the lattice is checked by a standard Monte Carlo procedure. Using very simple assumptions and a minimum number of parameters, photoexcitation curves and hysteresis loops under continuous irradiation below the thermal transition temperature can thus be simulated. The formation of clusters is analyzed and the presence of inhomogeneities in the system is investigated.
Cluster evolution in spin crossover systems observed in the frame of a mechano-elastic model
C. Enachescu, M. Nishino, S. Miyashita, L. Stoleriu, A. Stancu and A. Hauser Europhysics Letters, 91 (2) (2010), p27003 Keywords: Spin crossover, general studies of phase transitions, domain effects, magnetization curves, hysteresis
DOI:10.1209/0295-5075/91/27003 | unige:14746
In this paper we study the cluster formation and evolution in spin crossover systems during the thermal transition in the frame of a mechano-elastic model applied to open boundary hexagonal lattices. The switching processes between the high-spin (HS) and low-spin (LS) state are studied by a method combining a Monte Carlo standard procedure on the spin state and the lattice relaxation. In the present study, we adopt the transition probabilities of the spin state taking into account the energy gap between the two states, the degeneracy ratio and the local pressure determined by the elongations of the closest springs. It is found that clusters of molecules in the same state tend to grow starting from corners, as in available experimental data. Some qualitative differences between the processes of cluster formation for the two hysteresis branches, i.e., HS to LS and LS to HS are pointed out. Moreover, we have studied the dependence of cluster formation on the strength of the elastic interactions, and also on the system size. The size dependence of the ratio between the system size and the maximum cluster length is very weak, which indicates the appearance of macroscopic domains.
Effect of External Pressure on the Excitation Energy Transfer from [Cr(ox)3]3- to [Cr(bpy)3]3+ in [Rh1-xCrx(bpy)3][NaM1-yCry(ox)3]ClO4
M. Milos, P. Pal and A. Hauser ChemPhysChem, 11 (14) (2010), p3161-3166 Keywords: [Cr(bpy)3]3+; [Cr(ox)3]3−; 3D oxalate networks; excitation energy transfer; high pressures
DOI:10.1002/cphc.201000324 | unige:14715 | Abstract | Article HTML | Article PDF
Resonant excitation energy transfer from [Cr(ox)3]3- to [Cr(bpy)3]3+ in the doped 3D oxalate networks [Rh1-xCrx(bpy)3][NaMIII1-yCry(ox)3]ClO4 (ox=C2O4-, bpy=2,2â-bipyridine, M=Al,Rh) is due to two types of interaction, namely super exchange coupling and electric dipoleâdipole interaction. The energy transfer probability for both mechanisms is proportional to the spectral overlap of the 2Eâ4A2 emission of the [Cr(ox)3]3- donor and the 4A2â2T1 absorption of the [Cr(bpy)3]3+ acceptor.The spin-flip transitions of (pseudo-)octahedral Cr3+ are known to shift to lower energy with increasing pressure. Because the shift rates of the two transitions in question differ, the spectral overlap between the donor emission and the acceptor absorption is a function of applied pressure. For [Rh1-xCrx(bpy)3][Na-M1-yCry(ox)3]ClO4 the spectral overlap is thus substantially reduced on increasing pressure from 0 to 2.5 GPa. As a result, the energy transfer probability decreases with increasing pressure as evidenced by a decrease in the relative emission intensity from the [Cr(bpy)3]3+ acceptor.
Dynamic Perspective on the Function of Thermoresponsive Nanopores from in Situ AFM and ATR-IR Investigations
A.M. Popa, S. Angeloni, T. Bürgi, J.A. Hubbell, H. Heinzelmann and R. Pugin Langmuir, 26 (19) (2010), p15356-15365
DOI:10.1021/la102611k | unige:14783 | Abstract | Article HTML | Article PDF
This article describes the morphological and chemical characterization of stimuli-responsive functionalized silicon surfaces provided in parallel by atomic force spectroscopy (AFM) and Fourier transform infrared spectroscopy (FT-IR) enhanced by the single-beam sample reference attenuated total reflection method (SBSR-ATR). The stimuli-responsive behavior of the surfaces was obtained by grafting-to in melt carboxyl-terminated poly-N-isopropylacryl amides (PNIPAAM) with different degree of polymerization (DP) on epoxide-functionalized silicon substrates. The unprecedented real time and in situ physicochemical insight into the temperature-triggered response of the densely packed superficial brushes allowed for the selection of a PNIPAAM with a specific DP as a suitable polymer for the fabrication of silicon membranes exhibiting switchable nanopores. The fabrication process combines the manufacture of nanoporous silicon surfaces and their subsequent chemical functionalization by the grafting-to in melt of the selected polymer. Then, relevant information was obtained in what concerns the chemical modifications behind the topographical changes that drive the functioning of PNIPAAM-based hybrid nanovalves as well as the timescale on which the opening and closing of the nanopores occur.
Vibrational circular dichroism (VCD) spectra of small size-selected gold nanoparticles covered by both enantiomers of 1,1â²-binaphthyl-2,2â²-dithiol (BINAS) were measured. VCD spectra of particles covered by the opposite enantiomers of BINAS show a mirror image relationship. The VCD spectrum of adsorbed BINAS is different from the one of free BINAS and its disulfide form, but it resembles more the dithiol form. Detailed analysis reveals that the angle between the two binaphthyl rings of BINAS is close to 90° for the adsorbed BINAS, similar to what is found for the free molecule. VCD spectra are quite insensitive to the particles size, in contrast to the electronic CD spectra, which change drastically as the particle size increases. This indicates that the vibrational characteristic is a local property. A model of BINAS adsorbed on a Au10 cluster was used to calculate VCD spectra. As for free BINAS and the disulfide the calculated spectrum of the adsorbed BINAS is in very good agreement with the measured one. This shows the potential of VCD spectroscopy to gain insight into the conformation of chiral molecules adsorbed on small metal particles.
Pigments based on silica-coated gold nanorods: Synthesis, colouring strength, functionalisation, extrusion, thermal stability and colour evolution
C. Gautier, A. Cunningham, L. Si-Ahmed, G. Robert and T. Bürgi Gold Bulletin, 43 (2) (2010), p94-104 Keywords: gold, silica, nanorods, thermal stability, porosity, coloring strength
unige:14683 | Article PDF
The intense plasmon absorption bands of gold nanorods (GNRs) with peak extinction coefficients up to 6.4 x 109 M-1 cm-1 as well as their expected high stability make GNRs promising candidates for the colouration of bulk materials. The comparison of the integrated absorption in the visible region of GNRs with those of commercial organic pigments shows that the colouring strength of GNRs is 4 to 8 times higher. In order to improve their stability, GNRs were encapsulated in a silica shell of around 15 nm thickness using an optimized Stöber method. The silica surface was modified with octadecylsilane to enable their dispersion in non-polar media. Different plastics were successfully coloured with a tiny quantity of bare and functionalised GNRs@SiO2. These rods were homogeneously dispersed using extrusion. The shape of the rods was effectively stabilised by the silica shell at high temperature during the extrusion process. Surprisingly, a slight modification of the rods colour was observed due to a decrease of the refractive index in the mesoporous silica shell. However, this effect is greatly limited after the functionalisation.
The thermal conductivity of concentrated colloids in fluid, glass, and gel states was analyzed. SiO2 colloids at 10â31 vol % and Al2O3 colloids at 4.8 vol % in the fluid, the gel, and the glassy states were studied by dynamic light scattering, rheology, and transmission electron microscopy. Thermal conductivity of the three states was measured as a function of volume fraction. For the fluid and gel states the thermal conductivity increases almost linearly with concentration, reaching roughly 18% enhancement for silica at a volume fraction of 31 vol %. In contrast, in the glass state thermal conductivity strongly decreases with increasing volume fraction.
The photophysics and photochemistry of kynurenine (KN) covalently bound to the amino acids lysine, cysteine, and histidine, the antioxidant glutathione, and the protein lysozyme have been studied by optical spectroscopy with femto- and nanosecond time resolution. The fluorescence quantum yield of the adducts of KN to amino acids is approximately 2 times higher than that of the free KN in solution; KN attached to protein exhibits a 7-fold increase in the fluorescence quantum yield. The S1 state dynamics of KN-modified lysozyme reveals a multiphasic decay with a broad dispersion of time constants from 1 ps to 2 ns. An increase of the triplet yield of KN bound to lysozyme is also observed; the triplet state undergoes fast intramolecular decay. The obtained results reveal an increase of the photochemical activity of KN after its covalent attachment to amino acids and proteins, which may contribute to the development of oxidative stress in the human lensessthe main causative factor for the cataract onset.
Recent studies of organouranium chemistry have provided unusual pairs of similar polymetallic molecules containing (N)3â and (O)2â ligands, namely [(C5Me5)U(μ-I)2]3(μ3-N), 1, and [(C5Me5)U(μ-I)2]3(μ3-O), 2, and chair and boat conformations of [(C5Me5)2U(μ-N)U(μ-N3)(C5Me5)2]4, 3. These compounds were analyzed by density functional theory and multiconfigurational quantum chemical studies to differentiate nitride versus oxide in molecules for which the crystallographic data were not definitive and to provide insight into the electronic structure and unique chemical bonding of these polymetallic compounds. Calculations were also performed on [(C5Me5)2UN3(μ-N3)]3, 4, and [(C6F5)3BNU(N[Me]Ph)3], 5, for comparison with 1 and 3. On the basis of these results, the complex, [(C5Me5)U(μ3-E)]8, 6, for which only low-quality X-ray crystallographic data are available, was analyzed to predict if E is nitride or oxide.
Thermal Desorption, Vibrational Spectroscopic, and DFT Computational Studies of the Complex Manganese Borohydrides Mn(BH4)2 and [Mn(BH4)4]2−
G. Severa, H. Hagemann, M. Longhini, J.W. Kaminski, T.A. Wesolowski and C.M. Jensen Journal of Physical Chemistry C, 114 (36) (2010), p15516-15521
DOI:10.1021/jp101675q | unige:14754 | Abstract | Article HTML | Article PDF
The mechanochemical reaction of LiBH4 with MnCl2 produces the neutral complex Mn(BH4)2. Thermal desorption studies show that the mechanochemical reaction of NaBH4 with MnCl2produces a different species, apparently Na2Mn(BH4)4, that undergoes dehydrogenation of a much lower weight percent H at a ~20 °C higher temperature than the neutral Mn(BH4)2. Vibrational spectroscopy also reveals that a complex manganese borohydride(s) in addition to Mn(BH4)2 are formed from the mechanochemical reactions. Analysis of the vibrational spectra in conjunction with DFT calculations on a model Mn(BH4)42â complex suggest bidentate binding of the [BH4]â ligands to the Mn center in the anionic complex. The calculated highest frequencies of the BâH stretching modes (corresponding to the âfreeâ BâH bonds) agree well with the experimental frequencies and support the presence of this structural feature.
A series of bis(TTF) donors containing aromatic linkers between the two TTF units has been synthesized in order to investigate on the electronic structure of the oxidized species from an experimental and theoretical point of view. A mono(TTF)-pyridine compound has been also prepared and characterized by single-crystal X-ray diffraction analysis. Oxidation of a solution of 2,6-bis(TTF)-pyridine (TTF-Pyr-TTF) or of 1,3-bis(TTF)-benzene (TTF-Bz-TTF) in CH2Cl2 with less than 0.1 equivalent of [Cp2Fe][PF6] gives rise to a seven-line EPR spectrum consistent with the hyperfine structure calculated by DFT for the corresponding radical monocation. Increasing the proportion of oxidant leads to a four-line hyperfine structure, similar to the quartet pattern observed after oxidation of mono(TTF)-pyridine (Pyr-TTF) or mono(TTF)-benzene (Bz-TTF). In good accordance with the very weak value of J calculated by DFT for the dicationic biradicals these four-line spectra are attributed to [2,6-bis(TTF)-pyridine]2+ and [1,3-bis(TTF)-benzene]2+. Similar experimental results are obtained for 1,4-bis(TTF)-benzene. In this case, however, electrochemical oxidation leads to the monoradical at low potential and to the diradical at higher potential, while only the diradical could be observed by electrochemical oxidation of 2,6-bis(TTF)-pyridine or of 1,3-bis(TTF)-benzene.Â
Pressure and Temperature Influence on the Desorption Pathway of the LiBH4−MgH2 Composite System
U. Bösenberg, D.B. Ravnsbæk, H. Hagemann, V. D'Anna, C. Bonatto Minella, C. Pistidda, W. Van Beek, T.R. Jensen, R. Bormann and M. Dornheim Journal of Physical Chemistry C, 114 (35) (2010), p15212-15217
DOI:10.1021/jp104814u | unige:14742 | Abstract | Article HTML | Article PDF
The decomposition pathway in LiBH4âMgH2 reactive hydride composites was investigated systematically as a function of pressure and temperature. Individual decomposition of MgH2 and LiBH4 is observed at higher temperatures and low pressures (T ⥠450 °C and p(H2) ⤠3 bar), whereas simultaneous desorption of H2 from LiBH4 and formation of MgB2 was observed at 400 °C and a hydrogen backpressure of p(H2) = 5 bar. The simultaneous desorption of H2 from LiBH4 and MgH2 without intermediate formation of metallic Mg could not be observed. In situ X-ray diffraction (XRD) and infrared (IR) spectroscopy reveal the present crystalline and amorphous phases.
Zipper assembly of SHJ photosystems: focus on red naphthalenediimides, optoelectronic finetuning and topological matching
R. Bhosale, R.S.K. Kishore, V. Ravikumar, O. Kel, E. Vauthey, N. Sakai and S. Matile Chemical Science, 1 (2010), p357-368
DOI:10.1039/C0SC00177E | unige:14679 | Abstract | Article HTML | Article PDF
The objective of this study was to synthesize multichromophoric donor-acceptor systems with non-halogenated red (RO) naphthalenediimides (NDIs) attached along p-oligophenyl (POP) and oligophenylethynyl (OPE) scaffolds, and to evaluate their usefulness for zipper assembly of artificial photosystems. Compared to halogenated red NDIs (RCl, RBr), the HOMO of RO is 0.2 eV higher and the HOMO/LUMO gap 0.1 eV smaller, the latter introducing a shade of pink. Consistent with higher HOMO levels, RO zippers generate less photocurrent than RBr zippers in their respective action spectra. RO zippers are less sensitive to topological mismatch than RBr zippers and thus more robust and broadly applicable. Transient absorption measurements reveal efficient electron transfer from excited OPE donors to RO acceptors and less efficient hole injection from excited RO donors into OPE acceptors. Both processes demonstrate compatibility with OMARG-SHJ photosystems (supramolecular n/p-heterojunctions with oriented multicolored antiparallel redox gradients). Decreasing hole transfer with decreasing HOMO energy differences further demonstrates that SHJ-type hole injection disappears gradually (rather than abruptly). Losses in photonic energy during this process can thus be minimized by optoelectronic finetuning, but eventual gains in open circuit voltages risk coming with complementary losses in short circuit current.
The present study reports on the effect caused by sodium salts added to a solution of malachite green in a liquid/liquid interfacial system probed by the time-resolved surface second harmonic generation (TRSSHG) technique. This effect is known as âsalting-out effectâ and is shown to reveal two main issues: salts added to the bulk, first, cause a reduction of the dye solubility and, second, stimulate the adsorption of malachite green cations at the interface, changing the equilibrium constant between the dye molecules adsorbed at the interface and those being dissolved in the bulk. The increased adsorption at the interface is observed in the TRSSHG experiment as a relative increase of the aggregatesâ contribution to the measured time profile. However, depending on the nature and properties of salt anions, the mechanisms responsible for enhancing the population of interfacial aggregates can differ. This study explains such mechanisms for NaCl and NaSCN: addition of NaCl leads to an increase of the malachite green adsorption at the interface followed by the formation of aggregates, whereas the addition of NaSCN leads rather to the formation of aggregates already in the bulk with their further migration toward the interface. A simple quantitative description of the salting-out effect based on a modified Frumkin-Fowler-Guggenheim model also has been proposed. It has been shown to give a good agreement with the experiment with NaCl, i.e., when the formation of dye aggregates in the bulk solution can be neglected.
The properties of xanthurenic acid (XAN) in ground and photoexcited states have been studied using steady-state and time-resolved optical methods as well as quantum chemistry calculations. In neutral aqueous solution and in alcohols, XAN is present in a single tautomeric form (keto form), whereas in aprotic solvents and probably in basic aqueous solutions, more than one tautomeric form is present. UV irradiation of aqueous and alcoholic solutions of XAN results in a very rapid solvent-assisted tautomerization to the enol form, the later undergoes solvent-assisted transformation back to the keto form. The photolysis of XAN in aprotic solvents gives rise to the formation of numerous intermediate forms of XAN in both triplet and ground states. Under intense laser irradiation, XAN undergoes biphotonic ionization, the precursor for ionization being the excited singlet state.
The effect on crystal structure and vibrational frequencies of physical pressure in BaFCl and chemical pressure in Ba1âxSrxFCl solid solutions is studied using periodic density-functional theory (DFT) calculations performed within the local-density approximation (LDA) and the generalized gradient approximation (GGA). These results are compared with previously published experimental data for BaFCl in conjunction with new experimental data for Ba1âxSrxFCl and show overall a good agreement with experiment. The GGA method outperforms the LDA method for the description of BaFCl under pressure. However, the two DFT methods perform equally well for the description of the solid solutions, which have been studied within the virtual-crystal approximation. They also give consistent values of the energy of formation of Ba1âxSrxFCl, which can be correlated with the experimentally observed melting points. The comparison of the calculated mode Grüneisen parameters shows that, for the investigated systems, the effect of the chemical pressure and that of the physical pressure are not identical.
The correspondence between the exact embedding potential and the pair of the electron densitiesâthat of the embedded molecule and that of its environment [Wesolowski and Warshel, J. Phys. Chem.1993, 97, 8050]âis used to generate the average embedding potential and to subsequently calculate the solvatochromic shifts in a number of organic chromophores in solvents of various polarities. The averaged embedding potential is evaluated at a fictitious electron density of the solvent, which is obtained by means of âdressing upâ with electrons the classical site distributions derived from the statistical-mechanical, 3D molecular theory of solvation (aka 3D-RISM method) [Kovalenko In Molecular Theory of Solvation; Hirata, Ed.; Understanding Chemical Reactivity; 2003, Vol 24], self-consistently coupled with the electronic structure of the solute. The proposed approach to modeling solvatochromic shifts can be situated between the implicit and explicit type of models for the solvent. Numerical examples are given for the lowest-lying n â Ï* and Ï â Ï* excitations.
A series of pyridinium phenoxides that differ by the dihedral angle between the pyridinium and the phenoxide rings because of substituents with increasing steric encumbrance has been investigated by ultrafast spectroscopy. Like the related betaine-30, these molecules are characterised by a zwitterionic electronic ground state and a weakly polar S1Â state. Their fluorescence lifetime was found to lie between 200 to 750 fs, decreasing with increasing dihedral angle, and increasing with solvent viscosity. This was assigned to a non-radiative deactivation of the emissive state coupled to a large amplitude motion involving the dihedral angle. The transient absorption spectra suggested that emission occurs from the FranckâCondon S1Â state, which decays to a dark excited state, that itself most probably corresponds to the relaxed S1Â state. Finally, this relaxed state decays to the vibrationally hot ground state through an intramolecular charge separation process with a time constant ranging between 0.4 and 3 ps, increasing with the dihedral angle and with the solvent relaxation time. These variations were discussed in terms of the JortnerâBixon model of electron transfer, where the charge separation dynamics depends on both electronic coupling and solvent relaxation. The results suggested that charge separation slows down with increasing dihedral angle.
This feature article reviews research of core-substituted naphthalenediimides (cNDIs) in a comprehensive yet easily readable manner. Their synthesis, electrochemistry and spectroscopy are covered first with emphasis on the ability of cNDIs with electron donating substituents to absorb and fluoresce in all colors without global structural changes and cNDIs with electron withdrawing substituents to reach unprecedented extents of Ï-acidity. The section on supramolecular chemistry covers face-to-face Ï-stacks and peripheral hydrogen bonds, that on molecular recognition moves from pH and fluoride sensors to the binding to telomeric DNA in vivo and intercalation into Ï-stacks and sticky tweezers. cNDIs can recognize and transport anions by functional anionâÏ interactions. The section on electron transport describes cNDIs as air-stable n-semiconductors with high charge mobility and use as OFETs. Photoinduced electron transport by rainbow cNDIs has been used for the creation of artificial photosystems in solution, in bilayer membranes and on solid substrates. Examples include multicolor light harvesting architectures, organic solar cells, photosystems that can open up into ion channels, and supramolecular n/p-heterojunctions with antiparallel redox gradients. The review is highly interdisciplinary but should appeal most to organic, biosupramolecular and physical chemists.
The objective of this study was to evaluate the possibility of photoinduced stack/rod electron transfer in surface âzipperâ architectures composed of stacks of blue (B) naphthalenediimides (NDIs) along strings of oligophenylethynyl (OPE) rods. The synthesis and characterization of anionic and cationic multichromophoric OPE-B systems are reported. Absorption spectra suggest that in OPE-B systems, planarity and thus absorption and conductivity of the OPE can possibly be modulated by intramolecular stacking of the surrounding NDIs, although interfering contributions from aggregation remain to be differentiated. Among surface architectures constructed with OPE-B and POP-B systems by zipper and layer-by-layer (LBL) assembly, photocurrents generated by OPE-B zippers exhibit the best critical thickness and fill factors. These findings confirm the existence and functional relevance of topologically matching zipper architectures. In OPE-B zippers, OPEs generate much more photocurrent than the blue NDIs. Ultrafast electron transfer from OPEs to NDIs accounts for these photocurrents, providing wavelength-controlled access to rodâstack charge separation, and thus to formal supramolecular n/p-heterojunctions (SHJs). NDI excitation is not followed by the complementary hole transfer to the OPE rod. Scaffolds with higher HOMOs will be needed to integrate blue NDIs into SHJ photosystems.
Structural, Spectroscopic, and Multiconfigurational Quantum Chemical Investigations of the Electron-Rich Metal−Metal Triple-Bonded Tc2X4(PMe3)4 (X = Cl, Br) Complexes
F. Poineau, P.M. Forster, T.K. Todorova, L. Gagliardi, A.P. Sattelberger and K.R. Czerwinski Inorganic Chemistry, 49 (14) (2010), p6646-6654
DOI:10.1021/ic100641j | unige:14719 | Abstract | Article HTML | Article PDF
The compounds Tc2Cl4(PMe3)4 and Tc2Br4(PMe3)4 were formed from the reaction between (n-Bu4N)2Tc2X8 (X = Cl, Br) and trimethylphosphine. The Tc(II) dinuclear species were characterized by single-crystal XRD, UVâvisible spectroscopy, and cyclic voltammetry techniques, and the results are compared to those obtained from density functional theory and multiconfigurational (CASSCF/CASPT2) quantum chemical studies. The compound Tc2Cl4(PMe3)4 crystallizes in the monoclinic space group C2/c [a = 17.9995(9) à , b = 9.1821(5) à , c = 17.0090(9) à , β = 115.4530(10)°] and is isostructural to M2Cl4(PMe3)4 (M = Re, Mo, W) and to Tc2Br4(PMe3)4. The metalâmetal distance (2.1318(2) à ) is similar to the one found in Tc2Br4(PMe3)4 (2.1316(5) à ). The calculated molecular structures of the ground states are in excellent agreement with the structures determined experimentally. Calculations of effective bond orders for Tc2X82â and Tc2X4(PMe3)4 (X = Cl, Br) indicate stronger Ï bonds in the Tc24+ core than in Tc26+ core. The electronic spectra were recorded in benzene and show a series of low intensity bands in the range 10000â26000 cmâ1. Assignment of the bands as well as computing their excitation energies and intensities were performed at both TD-DFT and CASSCF/CASPT2 levels of theory. Calculations predict that the lowest energy band corresponds to the δ* â Ï* transition, the difference between calculated and experimental values being 228 cmâ1 for X = Cl and 866 cmâ1 for X = Br. The next bands are attributed to δ* â Ï*, δ â Ï*, and δ â Ï* transitions. The cyclic voltammograms exhibit two reversible waves and indicate that Tc2Br4(PMe3)4 exhibits more positive oxidation potentials than Tc2Cl4(PMe3)4. This phenomenon is discussed and ascribed to stronger metal (d) to halide (d) back bonding in the bromo complex. Further analysis indicates that Tc(II) dinuclear species containing Ï-acidic phosphines are more difficult to oxidize, and a correlation between oxidation potential and phosphine acidity was established.
Al3Li4(BH4)13: A Complex Double-Cation Borohydride with a New Structure
I. Lindemann, R.D. Ferrer, L. Dunsch, Y. Filinchuk, R. Cerný, H. Hagemann, V. D'Anna, L.M. Lawson Daku, L. Schultz and O. Gutfleisch Chemistry - A European Journal, 16 (2010), p8707-8712 Keywords: borohydrides; density functional calculations; hydrogen storage; metathesis; X-ray diffraction
DOI:10.1002/chem.201000831 | unige:14778 | Abstract | Article PDF
The new double-cation Al-Li-borohydride is an attractive candidate material for hydrogen storage due to a very low hydrogen desorption temperature (~70 °C) combined with a high hydrogen density (17.2 wt %). It was synthesised by high-energy ball milling of AlCl3 and LiBH4. The structure of the compound was determined from image-plate synchrotron powder diffraction supported by DFT calculations. The material shows a unique 3D framework structure within the borohydrides (space group=P-43n, a=11.3640(3) à ). The unexpected composition Al3Li4(BH4)13 can be rationalized on the basis of a complex cation [(BH4)Li4]3+ and a complex anion [Al(BH4)4]-. The refinements from synchrotron powder diffraction of different samples revealed the presence of limited amounts of chloride ions replacing the borohydride on one site. In situ Raman spectroscopy, differential scanning calorimetry (DSC), thermogravimetry (TG) and thermal desorption measurements were used to study the decomposition pathway of the compound. Al-Li-borohydride decomposes at ~70 °C, forming LiBH4. The high mass loss of about 20 % during the decomposition indicates the release of not only hydrogen but also diborane.
To study the reorientational motion of BH4 groups in the low-temperature (α) phase of Mg(BH4)2, we have performed nuclear magnetic resonance (NMR) measurements of the 1H and 11B spinâlattice relaxation rates in this compound over wide ranges of temperature (82â443 K) and resonance frequency (14â90 MHz for 1H and 14â28 MHz for 11B). It is found that the thermally activated reorientational motion in α-Mg(BH4)2 is characterized by a coexistence of at least three jump processes with strongly differing activation energies. Taking into account the anisotropy of the local environment of BH4 groups in α-Mg(BH4)2, these jump processes can be attributed to different types of reorientation. The nearly linear coordination of BH4 groups by two Mg atoms suggests that the fastest jump process corresponds to the rotation around the 2-fold axis connecting B and two Mg atoms, whereas the slowest process is associated with the rotation around two other 2-fold axes perpendicular to the MgâBâMg line.
The pressure/temperature phase diagram of LiAlH4 has been constructed by using Raman spectroscopy data. In situ high pressureâtemperature experiments were carried out using resistively heated diamond anvil cells up to 150 °C and 7 GPa. Room temperature phase transitions of monoclinic α-LiAlH4 â δ-LiAlH4 were observed at ~3.2 GPa. As the temperature is increased to ~100 °C, both the α and δ phases transform to β-LiAlH4 and remain stable up to 5.5 GPa. At temperatures greater than 300 °C, a new γ-LiAlH4 phase forms. Data of Konovalov (1995) has been used to define the phase boundary between β- and γ-LiAlH4 phases. We present a pressureâtemperature phase diagram of LiAlH4 based using diamond anvil cells coupled with Raman spectroscopy.
The mechanism of the photoinduced low-spin â high-spin spin crossover is actively being investigated in Fe(II) complexes in solution using ultrafast spectroscopies. These studies accurately inform on the reaction coordinate of the Fe(II) chromophore upon photoexcitation. However, they leave open questions regarding the role of the solvent. Here, we report the description from a fully ab initio molecular dynamics study of the structure of [Fe(bpy)3]2+ in water and of the organization of its solvation shell in the low-spin and the high-spin states. In particular, the low-spin â high-spin change of states is shown to be accompanied (i) by a 0.191 Ã lengthening of the FeâN bond, in agreement with experiment, and (ii) by an increased thermal fluctuation of the molecular edifice, which both result from the weakening of the FeâN bond. Furthermore, our results suggest that about two water molecules are expelled from the first solvation shell of [Fe(bpy)3]2+, which consists of water molecules intercalated between the bpy ligands.
Cation Size and Anion Anisotropy in Structural Chemistry of Metal Borohydrides. The Peculiar Pressure Evolution of RbBH4
Y. Filinchuk, A.V. Talyzin, H. Hagemann, V. Dmitriev, D. Chernyshov and B. Sundqvist Inorganic Chemistry, 49 (11) (2010), p5285-5292
DOI:10.1021/ic100359v | unige:14769 | Abstract | Article HTML | Article PDF
The pressure evolution of RbBH4 has been characterized by synchrotron powder X-ray diffraction and Raman spectroscopy up to 23 GPa. Diffraction experiments at ambient temperature reveal three phase transitions, at 3.0, 10.4, and 18 GPa (at 2.6, 7.8, and ~20 GPa from Raman data), at which the space group symmetry changes in the order Fm-3m(Z=4) âP4/nmm(2) âC222(2) âI-42m(4). Crystal structures and equations of state are reported for all four phases. The three high-pressure structure types are new in the crystal chemistry of borohydrides. RbBH4 polymorphs reveal high coordination numbers (CNs) for cation and anion sites, increasing with pressure from 6 to 8, via an intermediate 4 + 4 coordination. Different arrangements of the tetrahedral BH4 group in the Rb environment define the crystal symmetries of the RbBH4 polymorphs. The structural evolution in the MBH4 series is determined by the cationâs size, as it differs drastically for M = Li (CNs = 4, 6), Na (CN = 6), and Rb. The only structure common to the whole MBH4 family is the cubic one. Its bulk modulus linearly decreases as the ionic radius of M increases, indicating that the compressibility of the material is mainly determined by the repulsive BH4···BH4 interactions.
Deuteriumâhydrogen exchange in solid α-Mg(BH4)2 is demonstrated. Compared to the previously reported exchange reactions in the alkali borohydrides, the temperature at which isotope exchange starts to take place is significantly lower (132 °C vs 200 °C for LiBH4). The activation energy for the deuteriumâhydrogen exchange reaction is estimated to be 51 ± 15 kJ/mol. Almost complete isotope exchange was observed by treating solid Mg(BH4)2 for 72 h at 172 °C with 42 bar of D2. Preliminary experiments indicate that under these conditions Ca(BH4)2 also undergoes isotope exchange.
Inorganic borohydrides are actively studied in view of potential hydrogen storage applications. These compounds can be obtained by a variety of reactions ranging from high temperature reactions of the elements to exchange reactions in solution or in solid state. Different approaches will be discussed and compared.
Since the discovery of a formal quintuple bond in Arâ²CrCrArâ² (CrCr = 1.835 à ) by Power and co-workers in 2005, many efforts have been dedicated to isolating dichromium species featuring quintuple-bond character. In the present study we investigate the electronic configuration of several, recently synthesized dichromium species with ligands using nitrogen to coordinate the metal centers. The bimetallic bond distances of Powerâs compound and Cr2-diazadiene (1) (CrCr = 1.803 à ) are compared to those found for Cr2(μ-η2-ArNC(R)NAr)2 (2) (CrCr = 1.746 à ; R = H, Ar = 2,6-Et2C6H3), Cr2(μ-η2-ArXylNC(H)NArXyl)3 (3) (CrCr = 1.740reduced/1.817neutral à ; ArXyl= 2,6-C6H3-(CH3)2), Cr2(μ-η2-TippPyNMes)2 (4) (CrCr = 1.749 à ; TippPyNMes = 6-(2,4,6-triisopropylphenyl)pyridin-2-yl (2,4,6-trimethylphenyl)amide), and Cr2(μ-η2-DippNC(NMe2)N-Dipp)2 (5) (CrCr = 1.729 à , Dipp = 2,6-i-Pr2C6H3). We show that the correlation between the CrCr bond length and the effective bond order (EBO) is strongly affected by the nature of the ligand, as well as by the steric hindrance due to the ligand structure (e.g., the nature of the coordinating nitrogen). A linear correlation between the EBO and CrCr bond distance is established within the same group of ligands. As a result, the CrCr species based on the amidinate, aminopyridinate, and guanidinate ligands have bond patterns similar to the Arâ²CrCrArâ² compound. Unlike these latter species, the dichromium diazadiene complex is characterized by a different bonding pattern involving CrâNÏ interactions, resulting in a lower bond order associated with the short metalâmetal bond distance. In this case the short CrCr distance is most probably the result of the constraints imposed by the diazadiene ligand, implying a Cr2N4 core with a closer CrCr interaction.
Ab initio static and molecular dynamics study of the absorption spectra of the 4-styrylpyridine photoswitch in its cis and trans forms
L.M. Lawson Daku, J. Linares and M.-L. Boillot Physical Chemistry Chemical Physics, 12 (2010), p6107-6123
DOI:10.1039/b920850j | unige:13187 | Abstract
We report a thorough investigation of the absorption spectra of the cis and trans isomers of the 4-styrylpyridine photoswitch based on TDDFT calculations. The spectra of both isomers were analysed first from the results of excitation calculations performed on their optimised geometries. The main absorption band of the cis isomer is thus predicted to be due to the S0â S1 and S0â S2 transitions, while the main absorption band of the trans isomer is predicted to originate exclusively from the S0â S1 transition. The convolution of the calculated oscillator strengths with Gaussians helped mimic the broadening of the electronic transitions. However, it proved necessary to use Gaussians with a large full width at half maximum of 5000 cm-1; and, compared to experiment, the calculated main absorption bands of the two isomers are significantly red-shifted and far too symmetric. Consequently, as required for the detailed analysis of the finite-temperature absorption spectrum of a molecule as flexible as 4-styrylpyridine, the influence of the thermal fluctuations has been taken into account by calculating the spectra as time averages over CarâParrinello molecular dynamics trajectories. For both isomers, this led to a noticeable improvement in the relative positions of the calculated and experimental main absorption bands, and the asymmetry of the calculated bands brings them in better agreement with the experimental ones. Furthermore, these last results show that, actually, the S0â S1 and S0â S2 transitions both contribute significantly to the finite-temperature main absorption bands of the two isomers. Finally, in order to also take the vibrational broadening into account, the FranckâCondon factors of the relevant vibrations were calculated within the displaced harmonic oscillator approximation. By thus taking both the thermal and the vibrational broadening into account for the calculation of the absorption bands, the agreement between experiment and theory could be further improved.
Quantum chemical calculations were performed to investigate the cooperative effect of the nitrogen and silicon atoms on the singlet-triplet energy spacing and the reactivity of the singlet state in 1,2-diazacyclopentane-3,5-diyls and 1,2-diaza-4-silacyclopentane-3,5-diyls. The largest singlet-triplet energy gap (ÎEcST = -36.1 kcal/mol) found so far in localized 1,3-diradicals was in the C2v symmetry of 4,4-difluoro-1,2-diaza-4-silacyclopentane-3,5-diyl at the UB3LYP/6-31G(d) level of theory. The cooperative effect was also found in the energy differences of singlet diradicals with the corresponding ring-closing compounds, bicyclo[2.1.0]pentane derivatives. The singlet state of the 1,2-diaza-4-silacyclopentane-3,5-diyls was calculated to be energetically more stable than the ring-closing compound. The notable finding on the stability of the singlet diradicals may be attributed to the resonance structures that specifically stabilize the singlet state of diradicals. The computational studies predict that the singlet 1,2-diaza-4-silacyclopentane-3,5-diyl is a persistent molecule under conditions without intermolecular-trapping reagents.
The results of a computational study with multiconfigurational quantum chemical methods on actinide monoxides (AnO) and dioxides (AnO2) for An = Th, Pa, U, Np, Pu, Am, and Cm, are presented. First and second ionization energies were determined and compared with experimental values, when available. The trend along the series is analyzed in terms of the electronic configurations of the various species. The agreement with experiment is excellent in most cases. Of particular interest is the first ionization of PuO2. We applied cutting-edge theoretical methods to refine the ionization energy, but our computed data fall in the range of ~6 eV and not in the ~7 eV region as the experiment dictates. Such a system requires further computational and experimental attention.
Playing with a full deck: Single-crystal X-ray and neutron diffraction data show that the Th center in the title complex 1 (see structure; Th orange, B beige, N purple, C black, H blue) forms bonds with 15 H atoms, thus making 1 the first crystallographically characterized example of a complex with a Werner coordination number of fifteen. DFT calculations suggest that 1 adopts the fully symmetric 16-coordinate structure in the gas phase.
In the 3D network [Rh(bpy)3][NaCr(ox)3]ClO4 (ox = oxalate, bpy = 2,2'-bipyridine) phonon-assisted as well as resonant energy migration within the R1 line of the 4A2 â 2E transition of Cr3+ has been identified. The latter is dominant below 4.2 K, and in a fluorescence line narrowing spectrum, it manifests itself in a multiline pattern across the inhomogeneous line width with spacings corresponding to the zero-field splitting of the 4A2 ground state (Milos, M.; Kairouani, S.; Rabaste, S.; Hauser, A. Coord. Chem. Rev.2008, 252, 2540). H. Riesen demonstrated efficient spectral hole burning within the R1 line of Cr3+ doped at low concentrations into partially deuterated NaMg[Al(ox)3]·9H2O (Riesen, H. Coord. Chem. Rev.2006250, 1737). Here we show that at higher Cr3+ concentrations in the same host, both phenomena can be observed simultaneously, the resonant energy migration thus creating an additional series of persistent side holes.
The photophysics of aminoperylene (APe) in various solvents, including a room-temperature ionic liquid, has been investigated by steady-state and femtosecond transient absorption spectroscopies. The ultrafast excited-state dynamics originates from the solvation of the polar S1 state and not from a transition from a locally-excited to a charge-transfer state, as found with perylene-dimethylaniline. Addition of acid yields the protonated form APeH+, which exhibits similar photophysical properties than perylene, due to the suppression of the charge-transfer character of the S0âS1 transition. However, excited-state proton transfer, resulting to the formation of APe in the S1 state, is observed in methanol.
Structural Study of Mixed Crystals of [Zn1-xRux(bpy)3][NaCr(ox)3] Probed by High-Resolution Absorption Spectroscopy and High-Pressure Experiments
M. Milos, T. Penhouet, P. Pal and A. Hauser Inorganic Chemistry, 49 (7) (2010), p3402-3408
DOI:10.1021/ic902514w | unige:6468 | Abstract | Article HTML | Article PDF
In the mixed crystal series of the cubic three-dimensional networks of composition [Zn1âxRux(bpy)3][NaCr(ox)3] (0 ⤠x â¤1, ox = C2O42â, bpy = 2,2â²-bipyridine), high-resolution absorption spectroscopy in the region of the 4A2â2E transition (R-lines) reveals the creation of five specific spectroscopic sites for the [Cr(ox)3]3â complex. The concentration of these spectroscopic sites follows a binomial distribution of [Zn(bpy)3]2+ and [Ru(bpy)3]2+ among the four nearest neighbors of a given [Cr(ox)3]3â complex within the network. The tris-bipyridine complexes occupying those positions have an optimal ÏâÏ interaction with the oxalate ligands of the tris-oxalate chromophore. The energy of each spectroscopic [Cr(ox)3]3â site depends on the total concentration of [Ru(bpy)3]2+ in the mixed crystal and on its specific distribution among the four nearest neighbors. Single crystal X-ray diffraction indicates a reduction of the unit cell volume when [Zn(bpy)3]2+ (a = 15.6365(18) à ) is substituted by [Ru(bpy)3]2+ (a = 15.5098(6) à ). This alone would lead to a red-shift of the R lines in analogy to the red-shift of 25.2 cmâ1/GPa due to the decrease of the metal ligand CrâO bond length as observed in high-pressure luminescence experiments. However, specific ÏâÏ interactions with the nearest neighbors have the opposite effect and shift the transition in discrete jumps to higher energies with increasing [Ru(bpy)3]2+ mole fraction.
The electronic structure and the photophysical properties of the vanadium(III)ion in pseudo-octahedral oxygen coordination is reviewed. V3+ has received much interest from spectroscopists in recent years due to the advancement of state-of-the-art experimental techniques such as inelastic neutron scattering and high-field electron paramagnetic resonance spectroscopy that directly interrogate its large ground state zero-field splittings (ZFSs) and to rational parameterization of the ligand fieldp arameters using the angular overlap model. However, for V3+ these ZFSs can be large enough to also be probed directly by high-resolution electronic absorption spectroscopy of intra-configurational (t22g â t22g) spin-forbidden transitions in the near-IR and visible regions. The luminescent properties of V3+ with hexa-oxo and tris-bidentate di-oxo-coordination are quite disappointing compared to its neighbor in the periodic table, Cr3+, in similar environments. The efficient non-radiative pathways in these compounds are reviewed and compared to recent work on V3+ doped into NaMgAl(ox)3â 9H2O. The poor luminescence quantum efficiencies of V3+ oxo complexes is explained by strong coupling of multi-phonon processes with a dynamic Jahn-Teller distortion originating from the 3E trigonal component of the 3T1g ground state.
In a previous article we showed how to perform and analyze steady-state and nanosecond time-resolved experiments on fluorescence quenching by electron transfer in a coherent manner. Now, by making use of a superior time resolution, we explore the first stages of this kind of reaction. The novel information gained enables us to merge the results on the viscosity and the driving-force dependencies of the reaction rate. A unique set of parameters for a single reaction channel suffices to describe all the results in the frame of differential encounter theory for diffusion-influenced, bimolecular, remote electron-transfer reactions. The inclusion of the solvent structure is crucial for the understanding of the reaction kinetics. To the authors best knowledge, this is the first time that such a comprehensive set of data has been successfully and jointly explained in the field, with physically sound parameters for electron-transfer reactions.
Ultrafast Decay of the Excited Singlet States of Thioxanthone by Internal Conversion and Intersystem Crossing
G. Angulo, J. Grilj, E. Vauthey, L. Serrano-Andrés, O. Rubio-Pons and P. Jacques ChemPhysChem, 11 (2010), p480-488
DOI:10.1002/cphc.200900654 | unige:5125 | Abstract | Article HTML | Article PDF
The experimental ultrafast photophysics of thioxanthone in several aprotic organic solvents at room temperature is presented, measured using femtosecond transient absorption together with high-level ab initio CASPT2 calculations of the singlet- and triplet-state manifolds in the gas phase, including computed state minima and conical intersections, transition energies, oscillator strengths, and spin-orbit coupling terms. The initially populated singlet ÏÏ* state is shown to decay through internal conversion and intersystem crossing processes via intermediate nÏ* singlet and triplet states, respectively. Two easily accessible conical intersections explain the favorable internal conversion rates and low fluorescence quantum yields in nonpolar media. The presence of a singlet-triplet crossing near the singlet ÏÏ* minimum and the large spin-orbit coupling terms also rationalize the high intersystem crossing rates. A phenomenological kinetic scheme is proposed that accounts for the decrease in internal conversion and intersystem crossing (i.e. the very large experimental crescendo of the fluorescence quantum yield) with the increase of solvent polarity.
Effect of additives on the synthesis and reversibility of Ca(BH4)2
C. Rongeat, V. D'Anna, H. Hagemann, A. Borgschulte, A. Züttel, L. Schultz and O. Gutfleisch Journal of Alloys and Compounds, 493 (1-2) (2010), p281-287 Keywords: Hydrogen storage; calcium borohydride; reactive ball milling; Raman and infrared spectroscopy
DOI:10.1016/j.jallcom.2009.12.080 | unige:6412 | Abstract | Article PDF
Metal borohydrides are potential materials for solid state hydrogen due to their high gravimetric and volumetric hydrogen densities. Among them, Ca(BH4)2 is particularly interesting because of the predicted suitable thermodynamic properties. In this work, we investigate a new synthesis route using high pressure reactive ball milling. Starting from CaH2 and CaB6 with a TiCl3 or TiF3 as additive, a reaction yield of 19% is obtained after 24Â h milling at room temperature and 140Â bar H2. The presence of Ca(BH4)2 is confirmed by the presence of the stretching mode of the [BH4]- group in the infrared spectra of the as-milled samples. Using in-situ XRD, we observe the recrystallisation of a poorly crystallised Ca(BH4)2 phase present after milling. The reversible decomposition/formation of Ca(BH4)2 is obtained with higher yield (57%) using higher temperature and TiF3 as additive but not with TiCl3 despite its similar electronic structure. The differences observed using different additives and the influence of the anion are discussed.
NaSc(BH4)4: A Novel Scandium-Based Borohydride
R. Cerny, G. Severa, D.B. Ravnsbæk, Y. Filinchuk, V. D'Anna, H. Hagemann, D. Haase, C.M. Jensen and T.R. Jensen Journal of Physical Chemistry C, 114 (2010), p1357-1364 Keywords: Hydrogen storage material, crystal structure, vibrational spectra
DOI:10.1021/jp908397w | unige:4849 | Abstract | Article HTML | Article PDF
A new alkaline transition-metal borohydride, NaSc(BH4)4, is presented. The compound has been studied using a combination of in situ synchrotron radiation powder X-ray diffraction, thermal analysis, and vibrational and NMR spectroscopy. NaSc(BH4)4 forms at ambient conditions in ball-milled mixtures of sodium borohydride and ScCl3. A new ternary chloride Na3ScCl6 (P21/n, a = 6.7375(3) à , b = 7.1567(3) à , c = 9.9316(5) à , β = 90.491(3)°, V = 478.87(4) à 3), isostructural to Na3TiCl6, was identified as an additional phase in all samples. This indicates that the formation of NaSc(BH4)4 differs from a simple metathesis reaction, and the highest scandium borohydride yield (22 wt %) was obtained with a reactant ratio of ScCl3/NaBH4 of 1:2. NaSc(BH4)4 crystallizes in the orthorhombic crystal system with the space group symmetry Cmcm (a = 8.170(2) à , b = 11.875(3) à , c = 9.018(2) à , V = 874.9(3) à 3). The structure of NaSc(BH4)4 consists of isolated homoleptic scandium tetraborohydride anions, [Sc(BH4)4]â, located inside slightly distorted trigonal Na6 prisms (each second prism is empty, triangular angles of 55.5 and 69.1°). The experimental results show that each Sc3+ is tetrahedrally surrounded by four BH4 tetrahedra with a 12-fold coordination of H to Sc, while Na+ is surrounded by six BH4 tetrahedra in a quite regular octahedral coordination with a (6 + 12)-fold coordination of H to Na. The packing of Na+ cations and [Sc(BH4)4]â anions in NaSc(BH4)4 is a deformation variant of the hexagonal NiAs structure type. NaSc(BH4)4 is stable from RT up to â¼410 K, where the compound melts and then releases hydrogen in two rapidly occurring steps between 440 and 490 K and 495 and 540 K. Thermal expansion of NaSc(BH4)4 between RT and 408 K is anisotropic, and lattice parameter b shows strong anomaly close to the melting temperature.
The excited-state dynamics of five derivatives of the GFP-chromophore, which differ by the position and nature of their substituents, has been investigated in solvents of various viscosity and polarity and in rigid media using femtosecond-resolved spectroscopy. In polar solvents of low viscosity, like acetonitrile or methanol, the fluorescence decays of all compounds are multiexponential, with average lifetimes of the order of a few picoseconds, whereas in rigid matrices (polymer films and low temperature glasses), they are single exponential with lifetimes of the order of a few nanoseconds and fluorescence quantum yields close to unity. Global analysis of the fluorescence decays recorded at several wavelengths and of the transient absorption spectra reveals the presence of several excited-state populations with slightly different fluorescence and absorption spectra and with distinct lifetimes. These populations are attributed to the existence of multiple ground-state conformers. From the analysis of the dependence of the excited-state dynamics on the solvent and on the nature of the substituents, it follows that the nonradiative deactivation of all these excited chromophores involves an intramolecular coordinate with large amplitude motion. However, depending on the solvent and substituent, additional channels, namely, inter- and intramolecular hydrogen bond assisted nonradiative deactivation, are operative. This allows tuning of the excited-state lifetime of the chromophore. Finally, an ultrafast photoinduced intramolecular charge transfer is observed in polar solvents with one derivative bearing a dimethylaminophenyl substituent.
An Electrochemical and Photophysical Study of a Covalently Linked Inorganic–Organic Dyad
A. Kahnt, L.-P. Heiniger, S.-X. Liu, X. Tu, Z. Zheng, A. Hauser, S. Decurtins and D.M. Guldi ChemPhysChem, 11 (2010), p651-658 Keywords: cluster compounds, cyclic voltammetry, energy transfer, fullerenes, radiation chemistry
DOI:10.1002/cphc.200900728 | unige:6465 | Abstract | Article PDF
A molecular donor-acceptor dyad comprising a hexarhenium cluster core, [Re6(μ3-Se)8]2+, and a fullerene moiety which are covalently linked through a pyridine ligand was synthesized and fully characterized. The electrochemical and photophysical properties are reported. The detailed study includes cyclic voltammetry, steady-state absorption and fluorescence spectroscopy, radiation chemistry and transient absorption spectroscopy. A light-induced electron transfer between the inorganic cluster moiety and the fullerene can be excluded. However, a light-induced energy transfer from the rhenium cluster to the fullerene is proposed.









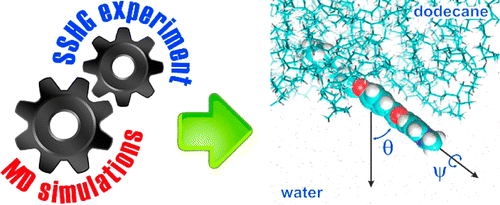


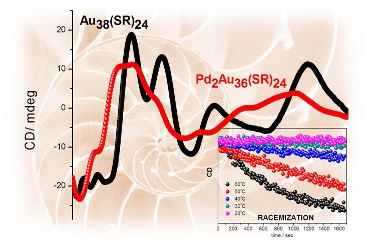

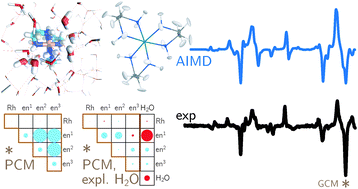


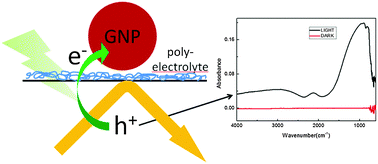
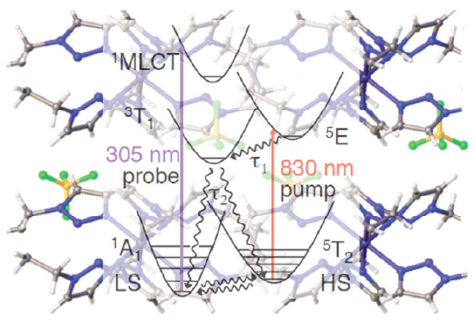





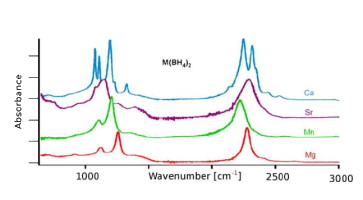
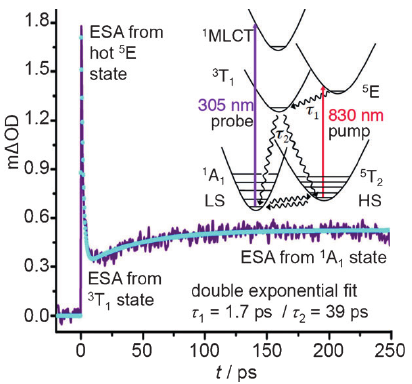

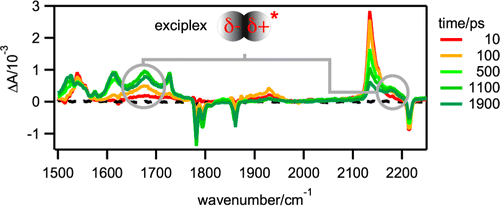


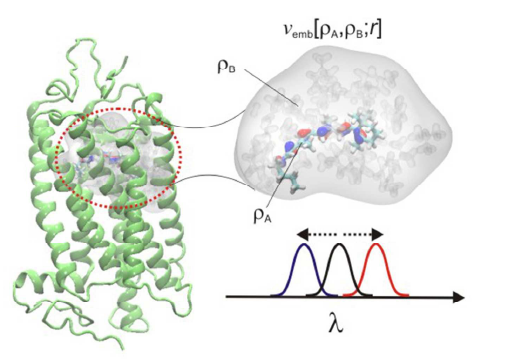
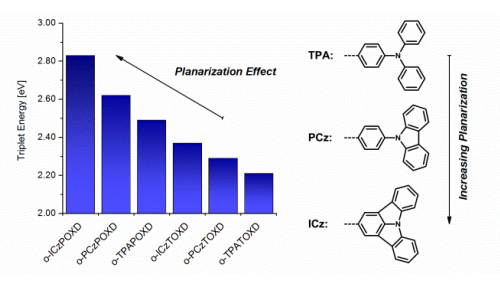
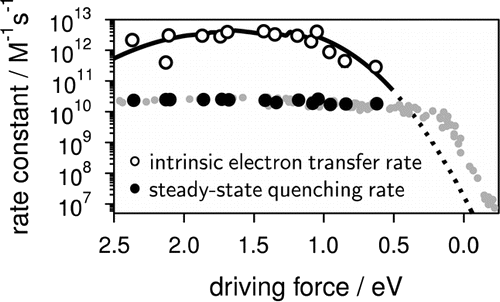
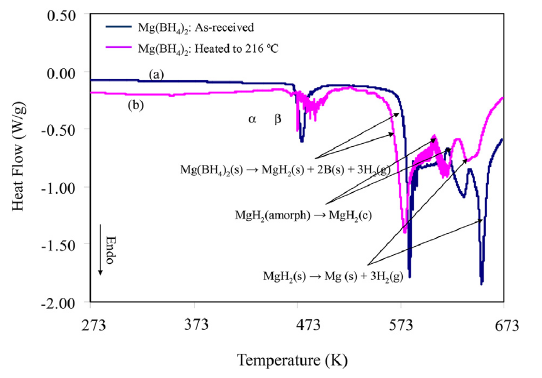
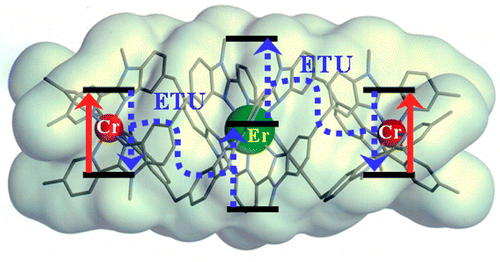

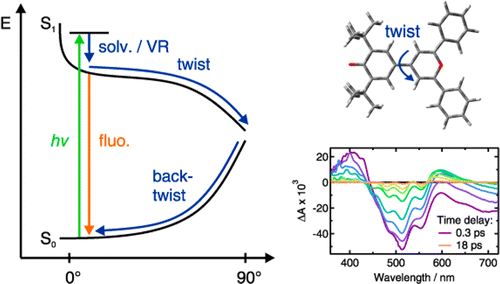



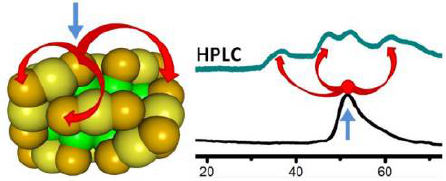
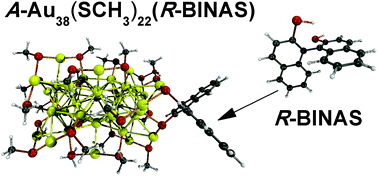

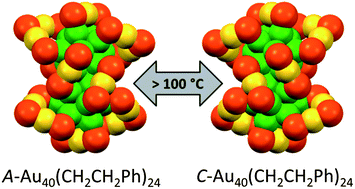
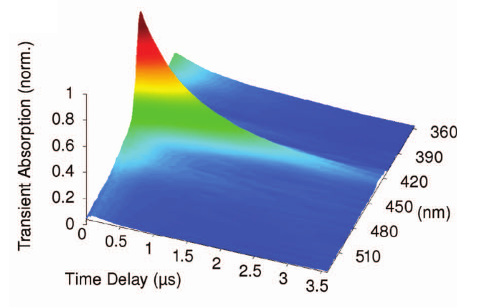
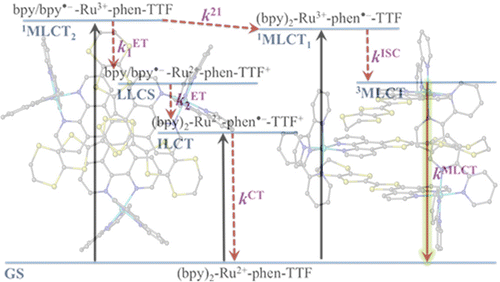
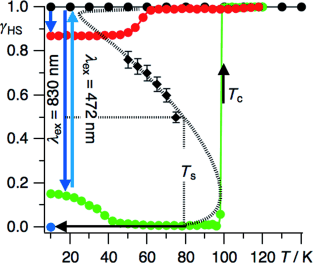


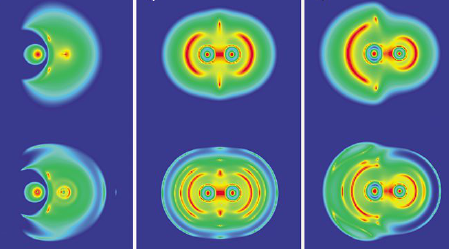

')){kind=link}
){kind=link}


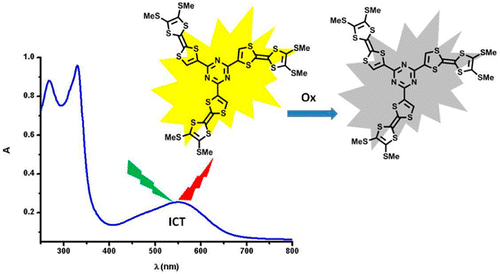



){kind=link}




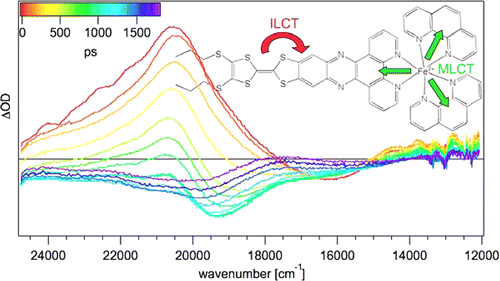
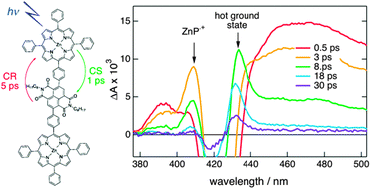
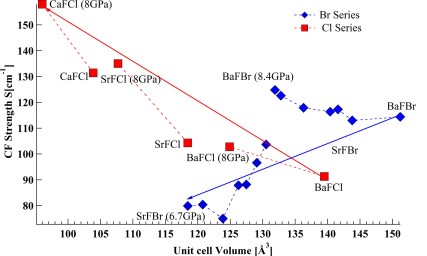

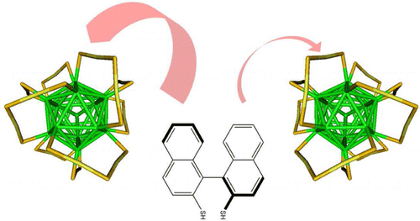
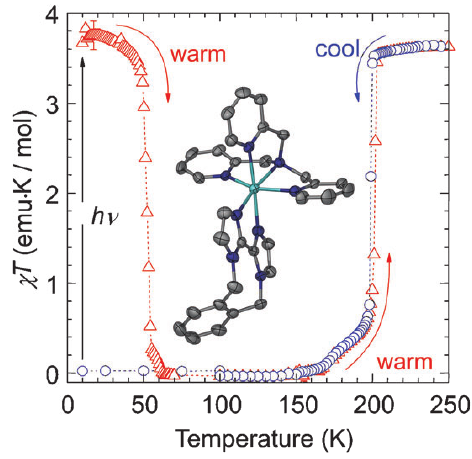

){kind=link}
){kind=link}


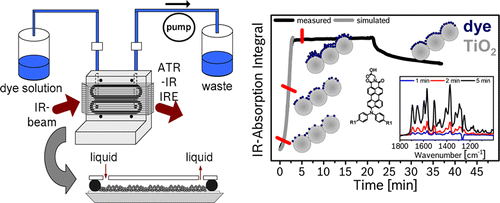
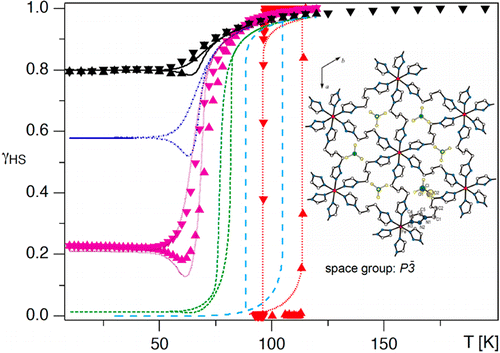



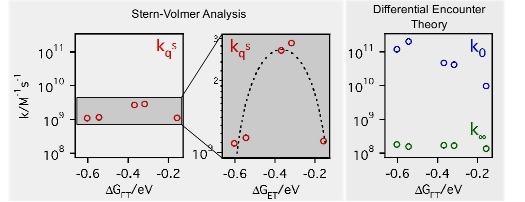
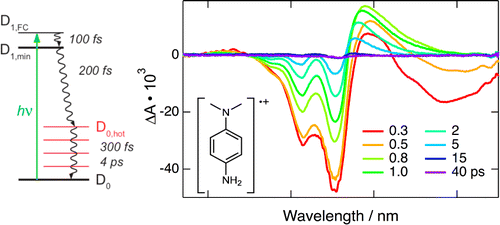


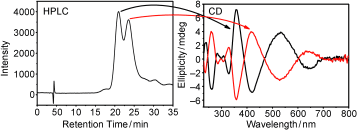
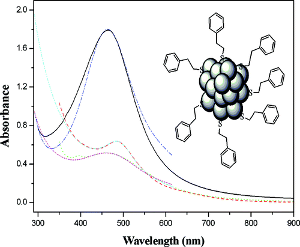

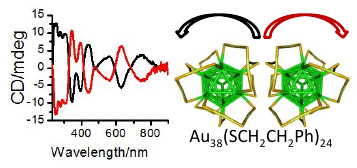


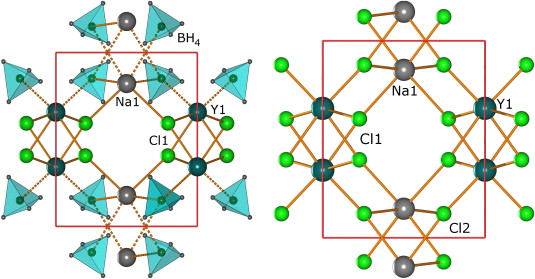





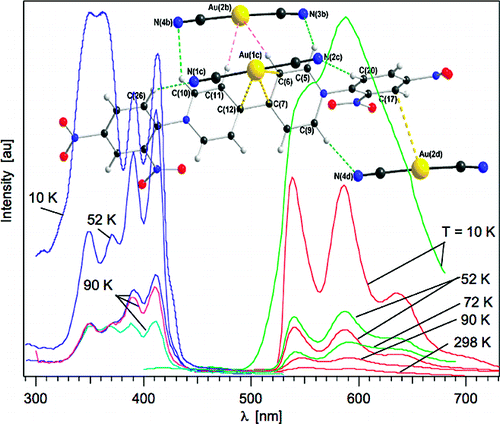
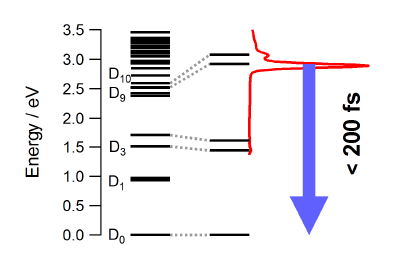

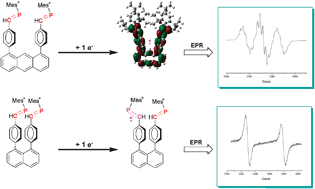

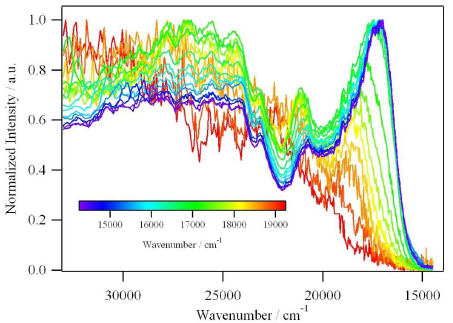
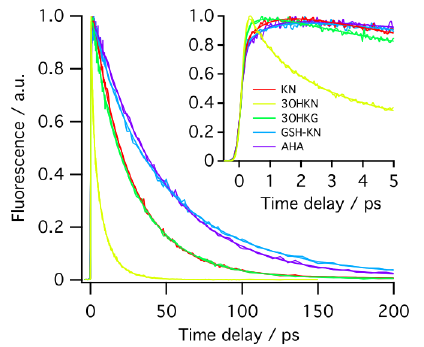

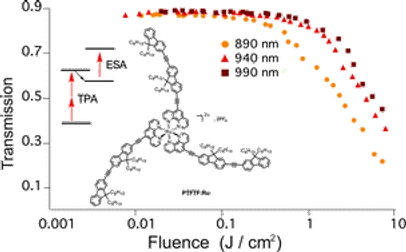
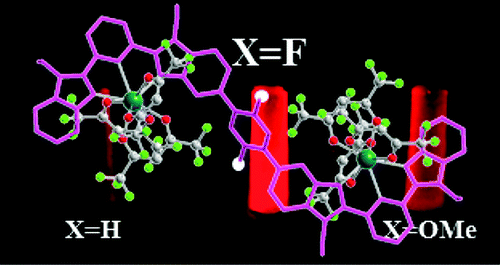
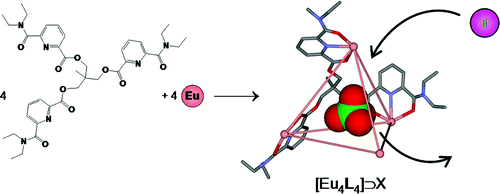
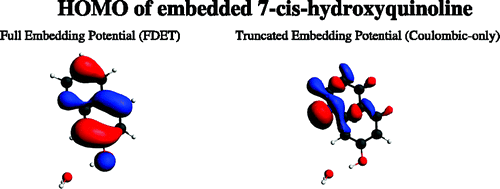




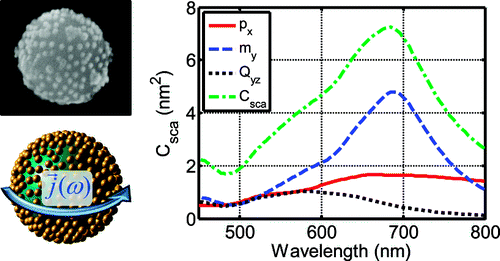

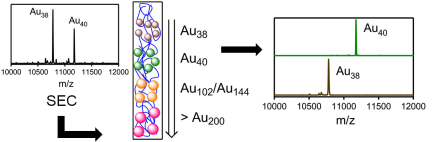
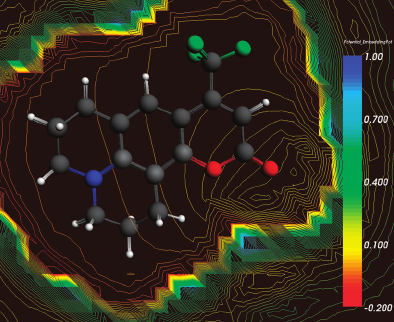


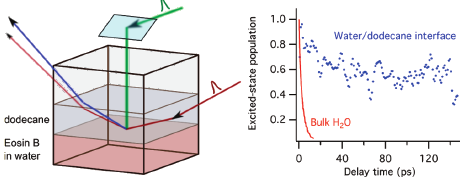
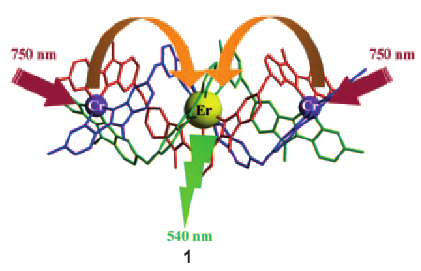
){kind=link}
){kind=link}
){kind=link}
){kind=link}
){kind=link}