Epigenetic Control of Developmental Processes

The normal morphogenesis of organs and structures requires a precise control of gene expression in time and in space during embryogenesis. The molecular processes that orchestrate these genetic programs are thus essential for ontogenesis, and their perturbations can lead to malformations or diseases. In this context, our laboratory focus on two main lines of research that synergistically aim at characterizing normal and pathological chromatin landscapes and their relevance toward morphogenesis.
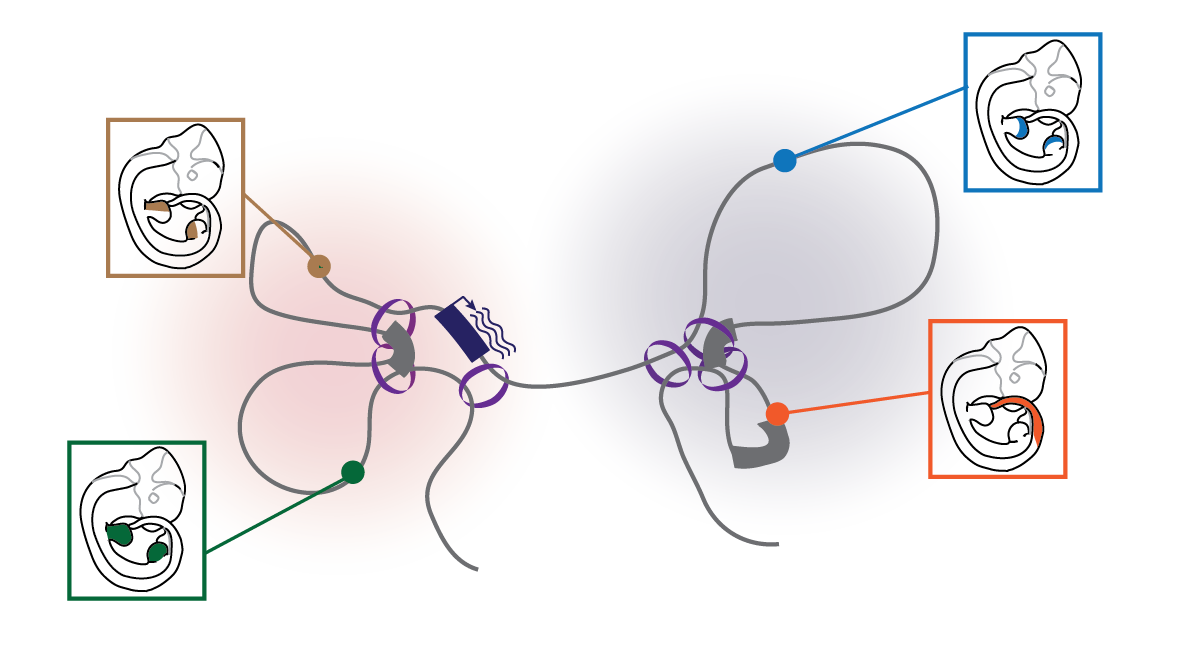
Developmental genes are often regulated by several enhancers (colored circles) with differential activities. In order to communicate with each other, enhancers and genes need to be in close spatial proximity.
Chromatin topology, gene regulatory control and morphogenesis
An important molecular process supporting gene transcriptional control is the communication, in the nuclear space, between gene regulatory regions (i.e. enhancers) and gene promoters. This communication is propagated through the 3D architecture of the genome, specifically through chromatin interactions, that can either be tissue-invariant or tissue-specific. In the latter case, the occurrences of interactions associate with the activities of genes and with changes of histone modifications at enhancers and promoters. Yet, it remains unknown if the formation and release of these interactions constitute a passive byproduct of gene activities or an active driver controlling the onset and offset of gene expression. To test the causal link between the deposition of histone marks or the formation of chromatin interactions in specific cell populations with changes in gene expression, we are developing targeted genetic and epigenetic engineering technologies, in vivo.
As an experimental model system, we use limb development and specific loci as testbed. On the one hand, we are expressing in vivo “activating” and “structural” dead Cas9 to alter chromatin modifications and topologies, respectively. On the other hand, we engineer mutations that force ectopic gene activity. We then apply chromatin technologies to survey the effect of these interventions on chromatin, gene transcription and morphogenesis. In summary, we aim at understanding the epigenetic events that drive the expression of genes during the development of complex organs.
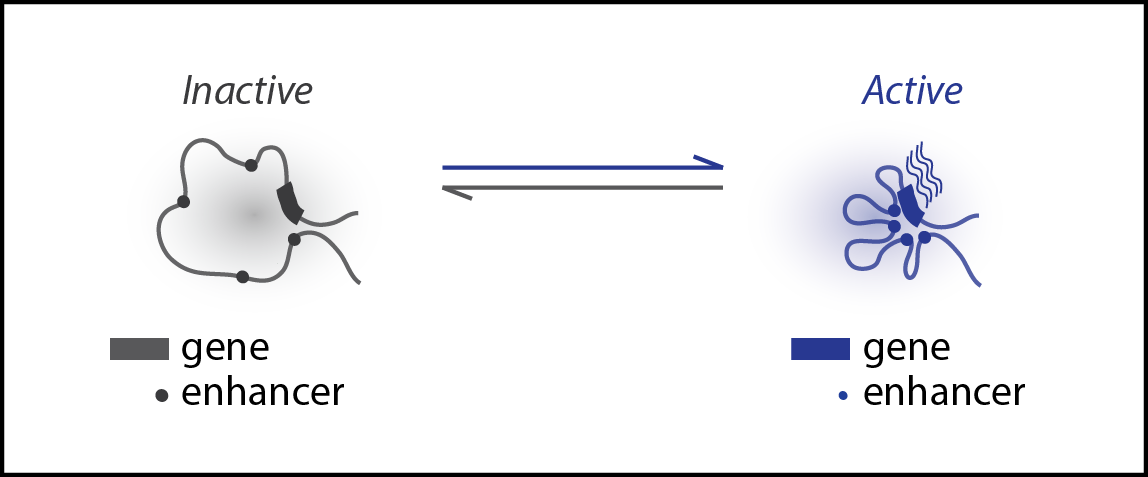
Enhancer-promoter chromatin interactions often correlate with the transcriptional activities of genes. Yet, it is unknown if these interactions are a passive byproduct of gene activities or an active driver controlling the onset and offset of gene expression
Dissecting chromatin biology in cell populations from which organ malformations originate
The transcriptional and chromatin-associated effects of mutations towards developing tissues in human patients and in mouse models is so far assessed via bulk quantification. Although this type of approach produces an averaged picture of the transcriptional, chromatin and structural alterations undergoing in the whole tissue, it lacks the cellular resolution necessary to understand where, when and how the mutations induce their pathogenic effect.
Here, we use on the one hand single cell RNA-seq approaches to profile the effect of re-engineered human deleterious mutations during mouse limb development. On the other hand, we use cell-sorting approaches to purify cells with altered gene expression in order to profile their chromatin landscapes and 3D genome structure. The combination of these approaches will determine the precise cellular and chromatin origins of transcriptional alterations taking place at cell population level within the developing limb and from which malformations result.
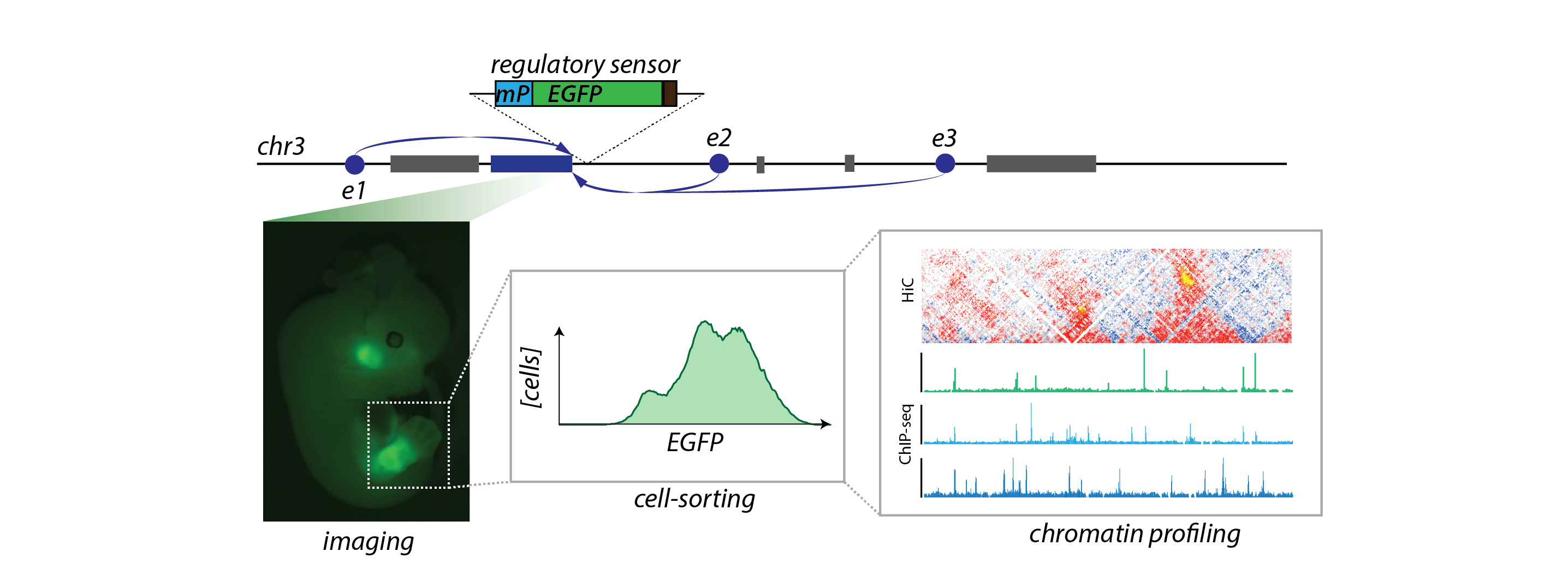
Regulatory sensors allow for the marking of cells with "active" regulatory landscapes. Sorting of fluorescent cells enable for the characterization of their chromatin structure.