

« Back to all publications
Download this list in a RIS file or a BIB file or a PDF file
|
||||||||
Ultrafast infrared transient absorption spectroscopy is used to study the photoinduced bimolecular electron transfer reaction between perylene in the first singlet excited state and 1,4-dicyanobenzene in acetonitrile and dichloromethane. Following vibrational marker modes on both donor and acceptor sides in real time provides direct insight into the structural dynamics during the reaction. A band narrowing on a time scale of a few tens of picoseconds observed on the antisymmetric CN stretching vibration of the dicyanobenzene radical anion indicates that a substantial part of the excess energy is channeled into vibrational modes of the product, despite the fact that the reaction is weakly exergonic. An additional narrowing of the same band on a time scale of several hundreds of picoseconds observed in acetonitrile only is interpreted as a signature of the dissociation of the geminate ion pairs into free ions. | ||||||||



|
|
||||||||
Luminophors of the perylene series containing the N,N-dimethylaniline residue in their molecules have been synthesised for the first time; spectral and luminescent properties of these compounds have been studied | ||||||||
|
 |
|||||||
A series of 6-styryl-2,4-diphenylpyrylium salts exhibiting dual fluorescence has been investigated by fluorescence up-conversion in conjunction with quantum chemical calculations. The short-wavelength emission is due to an excited state localized on the pyrylium fragment and the long-wavelength emission arises from a charge-transfer state delocalized over the whole molecule. The two fluorescing states do not exhibit a precursor−successor relationship. The rise time of the short-wavelength fluorescence is smaller than 200 fs, and that of the long-wavelength emission depends on the electron-donating property of the styryl group substituent. The rise is almost prompt with the weaker donors but is slower, wavelength and viscosity dependent with the strongest electron-donating group. A model involving a S2/S1 conical intersection is proposed to account for these observations. | ||||||||
|
||||||||
Femtosecond and nanosecond laser flash photolysis was used to determine the photophysical and photochemical processes in aqueous solutions of Fe(III) ion and 5-sulfosalicylic acid (SSA) containing the FeSSA complex and the free ligand. Excitation of the FeSSA complex in the charge transfer band (λmax = 505 nm) is followed by an ultrafast relaxation to the ground electronic state with two characteristic times of 0.26 and 1.8 ps. The shorter time constant is ascribed to internal conversion to the vibrationally hot electronic ground state of FeSSA and the 1.8 ps time constant is assigned to the vibrational cooling of the ground state. The UV irradiation of the solution (308 nm) leads to the excitation of both the free ligand and the FeSSA complex. The latter relaxes rapidly and the free ligand undergoes intersystem crossing to the triplet state. This system undergoes an irreversible photochemical reaction originating from an electron transfer (k = (9 ± 2) × 108 M−1 s−1) from the free ligand in the triplet state to the FeSSA complex. This electron transfer is accompanied by an energy transfer between these species (k = (1.3 ± 0.2) × 109 M−1 s−1). | ||||||||
|
||||||||
Rigid p-octiphenyl rods were used to create helical tetrameric π-stacks of blue, red-fluorescent naphthalene diimides that can span lipid bilayer membranes. In lipid vesicles containing quinone as electron acceptors and surrounded by ethylenediaminetetraacetic acid as hole acceptors, transmembrane proton gradients arose through quinone reduction upon excitation with visible light. Quantitative ultrafast and relatively long-lived charge separation was confirmed as the origin of photosynthetic activity by femtosecond fluorescence and transient absorption spectroscopy. Supramolecular self-organization was essential in that photoactivity was lost upon rod shortening (from p-octiphenyl to biphenyl) and chromophore expansion (from naphthalene diimide to perylene diimide). Ligand intercalation transformed the photoactive scaffolds into ion channels. | ||||||||
|
||||||||
Nanosecond laser flash photolysis, absorption and fluorescent spectroscopy were used to study the influence of pH on the photophysical and photochemical processes of 5-sulfosalicylic acid (SSA) in aqueous solutions. Information on the excited singlet state intramolecular proton transfer (ESIPT) of the SSA ions could be deduced from the dependence of the quantum yield and the spectral maximum of SSA fluorescence on the pH of the medium. The main photochemical active form of SSA at pH < 10 is the dianion (HSSA2−). Excitation of this species gives rise to the HSSA2− triplet state, to the SSA•2− radical anion and to the hydrated electron. In a neutral medium, the main decay channels of these intermediates are T–T annihilation, recombination and capture by the HSSA2− dianion, respectively. A decrease of pH leads to an increase of the second-order rate constants of disappearance of both HSSA2− triplet state and SSA•2− radical anion due to their protonation. | ||||||||
|
||||||||
The ultrafast ground state recovery (GSR) dynamics of the radical cation of perylene, Pe•+, generated upon bimolecular photoinduced electron transfer in acetonitrile, has been investigated using pump−pump−probe spectroscopy. With 1,4-dicyanobenzene as electron acceptor, the free ion yield is substantial and the GSR dynamics of Pe•+ was found to depend on the time delay between the first and second pump pulses, Δt12, i.e., on the “age” of the ion. At short Δt12, the GSR dynamics is biphasic, and at Δt12 larger than about500 ps, it becomes exponential with a time constant around 3 ps. With trans-1,2-dicyanoethylene as acceptor, the free ion yield is essentially zero and the GSR dynamics of Pe•+ remains biphasic independently of Δt12. The change of dynamics observed with 1,4-dicyanobenzene is ascribed to the transition from paired to free solvated ion, because in the pair, the excited ion has an additional decay channel to the ground state, i.e., charge recombination followed by charge separation. The rate constants deduced from the analysis of these GSR dynamics are all fully consistent with this hypothesis. | ||||||||
 |
|
|||||||
The excited-state dynamics of the DNA bisintercalator YOYO-1 and of two derivatives has been investigated using ultrafast fluorescence up-conversion and time-correlated single photon counting. The free dyes in water exist in two forms: nonaggregated dyes and intramolecular H-type aggregates, the latter form being only very weakly fluorescent because of excitonic interaction. The excited-state dynamics of the nonaggregated dyes is dominated by a nonradiative decay with a time constant of the order of 5 ps associated with large amplitude motion around the monomethine bridge of the cyanine chromophores. The strong fluorescence enhancement observed upon binding of the dyes to DNA is due to both the inhibition of this nonradiative deactivation of the nonaggregated dyes and the dissociation of the aggregates and thus to the disruption of the excitonic interaction. However, the interaction between the two chromophoric moieties in DNA is sufficient to enable ultrafast hopping of the excitation energy as revealed by the decay of the fluorescence anisotropy. Finally, these dyes act as solvation probes since a dynamic fluorescence Stokes shift was observed both in bulk water and in DNA. Very similar time scales were found in bulk water and in DNA. | ||||||||
|
 |
|||||||
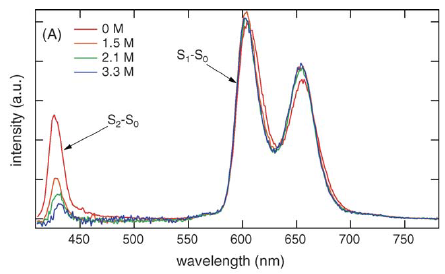
The dynamic Stokes shift of coumarin 153 has been measured in two room-temperature ionic liquids, 1-(3-cyanopropyl)-3-methylimidazolium bis(trifluoromethylsulfonyl)imide and 1-propyl-3-methylimidazolium tetrafluoroborate, using the fluorescence up-conversion technique with a 230 fs instrumental response function. A component of about 10−15% of the total solvation shift is found to take place on an ultrafast time scale < 10 ps. The amplitude of this component is substantially less than assumed previously by other authors. The origin of the difference in findings could be partly due to chromophore-internal conformational changes on the ultrafast time scale, superimposed to solvation-relaxation, or due to conformational changes of the chromophore ground state in polar and apolar environments. First three-pulse photon-echo peak-shift experiments on indocyanine green in room-temperature ionic liquids and in ethanol indicate a difference in the inertial component of the early solvent relaxation of <100 fs. | ||||||||
 |
|
|||||||
Several controversial questions in the field of bimolecular photoinduced electron transfer reactions in polar solvents are first briefly reviewed. Results obtained in our group using ultrafast spectroscopy and giving a new insight into these problems will then be described. They concern the driving force dependence of the charge separation distance, the formation of the reaction product in an electronic excited state, the absence of normal region for weakly exergonic charge recombination processes and the excitation wavelength dependence of the CR dynamics of donor–acceptor complexes. | ||||||||
Download this list in a RIS file or a BIB file or a PDF file
Contact:
Eric Vauthey
Physical Chemistry Department - Sciences II - University of Geneva
30, Quai Ernest Ansermet - CH-1211 Geneva 4 (Switzerland)
© All rights reserved by Eric Vauthey and the University of Geneva
Design and code by Guillaume Duvanel