

« Back to all publications
Download this list in a RIS file or a BIB file or a PDF file
|
 |
|||||||
The physicochemical properties of cationic dioxa (1), azaoxa (2), and diaza (3) [6]helicenes demonstrate a much higher chemical stability of the diaza adduct 3 (pKR+=20.4, Ered1/2 =â0.72â
V) compared to its azaoxa 2 (pKR+=15.2, Ered1/2=â0.45â
V) and dioxa 1 (pKR+=8.8, Ered1/2=â0.12â
V) analogues. The fluorescence of these cationic chromophores is established, and ranges from the orange to the far-red regions. From 1 to 3, a bathochromic shift of the lowest energy transitions (up to 614â
nm in acetonitrile) and an enhancement of the fluorescence quantum yields and lifetimes (up to 31â% and 9.8â
ns, respectively, at 658â
nm) are observed. The triplet quantum yields and circularly polarized luminescence are also reported. Finally, fine tuning of the optical properties of the diaza [6]helicene core is achieved through selective and orthogonal post-functionalization reactions (12â
examples, compounds 4â15). The electronic absorption is modulated from the orange to the far-red spectral range (560â731â
nm), and fluorescence is observed from 591 to 755â
nm with enhanced quantum efficiency up to 70â% (619â
nm). The influence of the peripheral auxochrome substituents is rationalized by first-principles calculations. | ||||||||



 |
|
|||||||
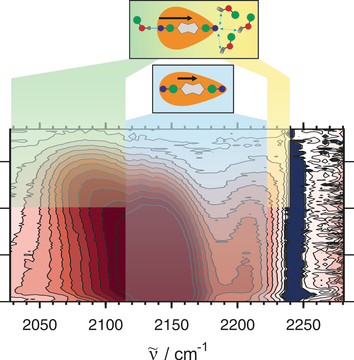
Symmetry-breaking charge transfer upon photoexcitation of a linear A-Ï-D-Ï-A molecule (D and A being electron donating and accepting groups) could be visualized using ultrafast time-resolved infrared spectroscopy by monitoring the CN stretching modes on the Aâ
units. Whereas in apolar solvents, the S1 state remains symmetric and quadrupolar, symmetry breaking occurs within ca. 100â
fs in polar solvents as shown by the presence of two CN bands, instead of one in apolar solvents, with a splitting that increases with polarity. In protic solvents, symmetry breaking is significantly amplified by H-bonding interactions, which are the strongest at the CN group with the highest basicity. In strongly protic solvents, the two CN bands transform in about 20â
ps into new bands with a larger splitting, and the lifetime of the S1 state is substantially reduced. This is attributed to the formation of an excited asymmetric tight H-bond complex. | ||||||||
|
|
 |
|||||||
The linear and nonlinear optical properties of a series of oligomeric squaraine dyes were investigated by one-photon absorption spectroscopy (1PA) and two-photon absorption (2PA) induced fluorescence spectroscopy. The superchromophores are based on two indolenine squaraine dyes with transoid (SQA) and cisoid configuration (SQB). Using these monomers, linear dimers and trimers as well as star-shaped trimers and hexamers with benzene or triphenylamine cores were synthesised and investigated. The red-shifted and intensified 1PA spectra of all superchromophores could well be explained by exciton coupling theory. In the linear chromophore arrangements we also found superradiance of fluorescence but not in the branched systems. Furthermore, the 2PA showed enhanced cross sections for the linear oligomers but only additivity for the branched systems. This emphasizes that the enhancement of the 2PA cross section in the linear arrangements is probably caused by orbital interactions of higher excited configurations. | ||||||||
 |
|
|||||||
Unprecendented regioselective post-functionalization of racemic and enantiopure cationic diaza [4]helicenes is afforded. The peripheral auxochrome substituents allow a general tuning of the electrochemical, photophysical and chiroptical properties of the helical dyes (26 examples). For instance, electronic absorption and circular dichroism are modulated from the orange to near-infrared spectral range (575-750 nm), fluorescence quantum efficiency is enhanced up to 0.55 (631 nm) and circularly polarized luminescence is recorded in the red (âglumâ ~ 10-3) | ||||||||


|
 |
|||||||
Most symmetric quadrupolar molecules designed for two-photon absorption behave as dipolar molecules in the S1Â electronic excited state. This is usually explained by a breakup of the symmetry in the excited state. However, the origin of this process and its dynamics are still not fully understood. Here, excited-state symmetry breaking in a quadrupolar molecule with a D-Ï-A-Ï-D motif, where D and A are electron donating and accepting units, is observed in real time using ultrafast transient infrared absorption spectroscopy. The nature of the relaxed S1Â state was found to strongly depend on the solvent polarity: (1) in nonpolar solvents, it is symmetric and quadrupolar; (2) in weakly polar media, the quadrupolar state observed directly after excitation transforms to a symmetry broken S1Â state with one arm bearing more excitation than the other; and (3) in highly polar solvents, the excited state evolves further to a purely dipolar S1Â state with the excitation localized entirely on one arm. The time scales associated with the transitions between these states coincide with those of solvation dynamics, indicating that symmetry breaking is governed by solvent fluctuations. | ||||||||
')){kind=link}
 |
|
|||||||
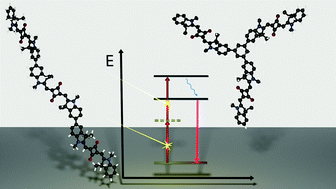
A molecular pentad comprised of a central multielectron donor and two flanking photosensitizer-acceptor moieties was prepared in order to explore the possibility of accumulating two positive charges at the central donor, using visible light as an energy input. Photoinduced charge accumulation in purely molecular systems without sacrificial reagents is challenging, because of the multitude of energy-wasting reaction pathways that are accessible after excitation with two photons. As expected, the main photoproduct in our pentad is a simple electronâhole pair, and it is tricky to identify the desired two-electron oxidation product on top of the stronger signal resulting from one-electron oxidation. | ||||||||
|
 |
|||||||
The properties of binary mixtures of dimethylsulfoxide and glycerol, measured by several techniques, are reported. Special attention is given to those properties contributing or affecting chemical reactions. In this respect the investigated mixture behaves as a relatively simple solvent and it is especially well suited for studies on the influence of viscosity in chemical reactivity. This is due to the relative invariance of the dielectric properties of the mixture. However, special caution must be taken with specific solvation, as the hydrogen-bonding properties of the solvent changes with the molar fraction of glycerol. | ||||||||
|
||||||||
Late endosomes are a major trafficking hub in the cell at the crossroads between endocytosis, autophagy, and degradation in lysosomes. Herein is disclosed the first small molecule allowing their selective imaging and monitoring in the form of a diazaoxatriangulene fluorophore, 1a (hexadecyl side chain). The compound is prepared in three steps from a simple carbenium precursor. In nanospheres, this pH-sensitive (pKa = 7.3), photochemically stable dye fluoresces in the red part of visible light (601 and 578 nm, acid and basic forms, respectively) with a quantum yield between 14 and 16% and an excited-state lifetime of 7.7â7.8 ns. Importantly, the protonated form 1a·H+ provokes a specific staining of late endosome compartments (pH 5.0â5.5) after 5 h of incubation with HeLa cells. Not surprisingly, this late endosome marking depends on the intra-organelle pH, and changing the nature of the lipophilic chain provokes a loss of selectivity. Interestingly, fixation of the fluorophore is readily achieved with paraformaldehyde, giving the possibility to image both live and fixed cells. | ||||||||
 |
|
|||||||
The properties of a series of oxazole yellow dyes, including the dicationic YOPRO-1 and its homodimeric parent YOYO-1 and two monocationic dyes (YOSAC-1 and YOSAC-3), have been investigated at the dodecane/water interface using stationary and time-resolved surface second harmonic generation (SSHG) combined with quantum chemical calculations. Whereas YOYO-1 exists predominantly as a H-dimer in aqueous solution, the stationary SSHG spectra reveal that such dimers are not formed at the interface. No significant H-aggregation was observed with YOPRO-1, neither in solution nor at the interface. In the case of the monocationic YOSAC dyes, a distinct SSHG band due to H-aggregates was measured at the interface, whereas only weak aggregation was found in solution. These distinct aggregation behaviors can be explained by the different orientations of the dyes at the interface, as revealed from the analysis of polarization-resolved experiments, the doubly-charged dyes lying more flat on the interface than the singly charged ones. Although YOYO-1 and YOPRO-1 do not form H-dimer/aggregates at the interface, time-resolved SSHG measurements point to the occurrence of intra- and intermolecular interactions, respectively, which inhibit the ultrafast non-radiative decay of the excited dyes via large amplitude motion, and lead to a nanosecond excited-state lifetime. The distinct behavior evidenced here for YOYO-1 and YOSAC dyes points to their potential use as fluorescent or SHG interfacial probes. | ||||||||
Download this list in a RIS file or a BIB file or a PDF file
Contact:
Eric Vauthey
Physical Chemistry Department - Sciences II - University of Geneva
30, Quai Ernest Ansermet - CH-1211 Geneva 4 (Switzerland)
© All rights reserved by Eric Vauthey and the University of Geneva
Design and code by Guillaume Duvanel