

« Back to all publications
Download this list in a RIS file or a BIB file or a PDF file
|
 |
|||||||
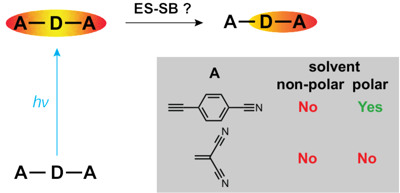
A significant number of quadrupolar dyes behave as their dipolar analogues when photoexcited in polar environments. This is due to the occurrence of excited-state symmetry breaking (ES-SB), upon which the electronic excitation, initially distributed over the whole molecule, localises preferentially on one side. Here, we investigate the ES-SB properties of two A–D–A dyes, consisting of a pyrrolo-pyrrole donor (D) and either cyanophenyl or dicyanovinyl acceptors (A). For this, we use time-resolved vibrational spectroscopy, comparing IR absorption and femtosecond stimulated Raman spectroscopies. Although dicyanovinyl is a stronger electron-withdrawing group, ES-SB is not observed with the dicyanovinyl-based dye even in highly polar media, whereas it already takes place in weakly polar solvents with dyes containing cyanophenyl accepting groups. This difference is attributed to the large electronic coupling between the D–A branches in the former dye, whose loss upon symmetry breaking cannot be counterbalanced by a gain in solvation energy. Comparison with analogues of the cyanophenyl-based dye containing different spacers reveals that interbranch coupling does not so much depend on the distance between the D–A subunits than on the nature of the spacer. We show that transient Raman spectra probe different modes of these centrosymmetric molecules but are consistent with the transient IR data. However, lifetime broadening of the Raman bands, probably due to the resonance enhancement, may limit the application of this technique for monitoring ES-SB. | ||||||||



 |
|
|||||||
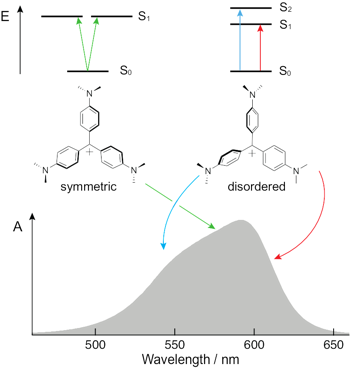
The nature of the lowest-energy electronic absorption band of crystal violet (CV) and particularly the origin of its high-energy shoulder have been debated since the middle of the past century. The most recent studies invoke a splitting of the S1 state upon symmetry breaking induced by interactions with the solvent and/or the counterion. Using a combination of stationary and time-resolved polarized spectroscopy together with quantum-chemical calculations, we show that torsional disorder in the ground-state results in an inhomogeneous broadening of the absorption band of CV. The center of the band is mostly due to symmetric molecules with a degenerate S1 state, whereas the edges originate from transitions to the S1 and S2 states of distorted symmetry-broken molecules. Transient-absorption measurements with different excitation wavelengths reveal that these two groups of molecules interconvert rapidly in liquid but not in a rigid environment. | ||||||||
|
 |
|||||||
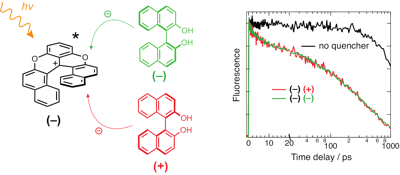
Occurrence of chiral recognition in bimolecular photoinduced electron transfer (ET) is difficult to identify because of the predominant role of diffusion. To circumvent this problem, we apply a combination of ultrafast time-resolved fluorescence and transient electronic absorption to look for stereoselectivity in the initial, static stage of ET quenching, where diffusion is not relevant. The fluorophore and electron acceptor is a cationic hexahelicene, whereas the quencher has either stereocentered (tryptophan) or axial (binaphthol) chirality. We found that, in all cases, the quenching dynamics are the same, within the limit of error, for different diastereomeric pairs in polar and medium-polar solvents. The same absence of chiral effect is observed for the recombination of the radical pair, which results from the quenching. Molecular dynamics simulations suggest that the distribution of inter-reactant distance is independent of the chirality of the acceptor and the donor. Close contact resulting in large electronic coupling is predicted to be possible with all diastereomeric pairs. In this case, ET is an adiabatic process, whose dynamics do no longer depend on the coupling, but are rather controlled by high-frequency intramolecular modes. | ||||||||
Download this list in a RIS file or a BIB file or a PDF file
Contact:
Eric Vauthey
Physical Chemistry Department - Sciences II - University of Geneva
30, Quai Ernest Ansermet - CH-1211 Geneva 4 (Switzerland)
© All rights reserved by Eric Vauthey and the University of Geneva
Design and code by Guillaume Duvanel