
|
Photophysics and Photochemistry of Transition Metal Compounds |
| Home Research Members Collaborations Publications |

Publications by Prof. Hagemann


2018
2017
2016
2015
2014
2013
2012
2011
2010
2009
2008
2007
2006
2005
2004
2002
2000
1999
1998
1997
1996
1995
1994
1993
1992
1991
1990
1989
1988
1987
1986
1985
1984
1982
|



|
||||||||
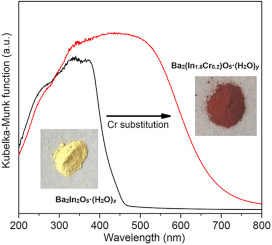
Cr-substituted polycrystalline Ba2(In2-xCrx)O5·(H2O)δ powders (0.04 ≤ x ≤ 0.60) were synthesized by solid state reaction to investigate the relation of crystal structure, thermochemical, magnetic, and optical properties. The Cr-substitution results in an unit cell expansion and formation of the higher-symmetric tetragonal phase together with increased oxygen and hydrogen contents. Magnetic property measurements reveal that the diamagnetic pristine Ba2In2O5·(H2O)δ becomes magnetically ordered upon Cr-substitution. By UV–vis spectroscopy a gradual shift of the absorption-edge energy to lower values was observed. Numerical calculations showed that the observed bandgap narrowing was ascribed to the Cr induced states near the Fermi level. The correlation between the changes of crystal chemistry, magnetic, and optical properties of Cr-substituted Ba2(In2-xCrx)O5·(H2O)δ can be explained by the replacement of In by Cr. Consequently, an enhanced photocatalytic CO2 reduction activity was observed with increasing Cr substitution, compatible with the state-of-the-art high surface area TiO2 photocatalyst (P-25). | ||||||||
|
|
 |
|||||||
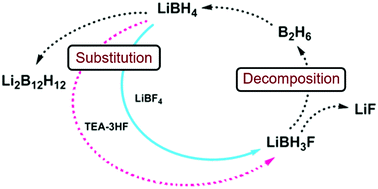
Fluoride substitution in LiBH4 is studied by investigation of LiBH4-LiBF4 mixtures (9:1 and 3:1). Decomposition was followed by in-situ synchrotron radiation X-ray diffraction (in-situ SR-PXD), thermogravimetric analysis and differential scanning calorimetry with gas analysis (TGA/DSC-MS) and in-situ infrared spectroscopy (in-situ FTIR). Upon heating, fluoride substituted LiBH4 forms (LiBH4-xFx) and decomposition occurs, releasing diborane and solid decomposition products. The decomposition temperature is reduced more than fourfold relative to the individual constituents, with decomposition commencing at T / °C = 80 °C. The degree of fluoride substitution is quantified by sequential Rietveld refinement and shows a selective manner of substitution. In-situ FTIR experiments reveal formation of bands originating from LiBH4-xFx. Formation of LiF and observation of diborane release implies that the decomposing materials have a composition that facilitates formation of diborane and LiF, i.e. LiBH4-xFx (LiBH3F). An alternative approach for fluoride substitution was performed, by addition of Et3N∙3HF to LiBH4, yielding extremely unstable products. Spontaneous decomposition indicates fluoride substitution to have occurred. From our point of view, this is the most significant destabilization effect seen for borohydride materials so far. | ||||||||
 |
|
|||||||
Cr-substituted and pristine Ba2In2O5·(H2O)x powders were synthesized by solid state reaction. The influence of Cr-substitution on the crystal structure, chemical composition, magnetic and optical properties were investigated. Powder X-ray diffraction (XRD), elemental analysis and TGA-MS reveal that with substitution of In for Cr, the unit cell volume and the unit cell parameter b increase together with the oxygen and hydrogen content. Magnetic property measurements indicate that Ba2In2O5·(H2O)x is diamagnetic in the temperature range of 2 K < T < 300 K becoming ferromagnetic upon Cr-substitution. In the UV–vis spectra of the Cr-substituted sample a distinctive shift of the absorption-edge energy from 430 to 690 nm was observed corresponding to a bandgap narrowing from 2.88 to 1.80 eV. The replacement of tetrahedral InO4 units by octahedral CrO6 units was found to be the main factor for the drastic change of the magnetic and optical properties. | ||||||||
|
 |
|||||||
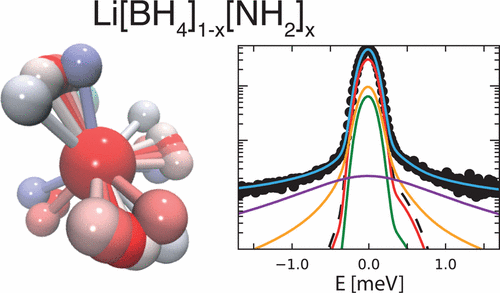
Lithium amide–borohydrides Li[BH4]1–x[NH2]x possess liquid-like Li superionic conductivity at nearly ambient temperature. The fast Li+ diffusion facilitated by the localized motions of the anions is proposed to occur through a network of vacant tetrahedral sites, acting as conduction channels. To study the reorientational dynamics of the anions, we have performed quasielastic neutron scattering experiments on samples with different compositions (x = 2/3, 0.722, 0.737, 3/4) over a broad temperature and time range. To unambiguously disentangle the contributions of the two species, [BH4]− and [NH2]−, we took advantage of deuterium labeling and could clearly demonstrate that the quasielastic broadening is mainly determined by the [BH4]− reorientations. With the help of a newly developed model, supported by ab initio molecular dynamics calculations, we have identified three relaxation components, which account for generally anisotropic C3-rotations of the [BH4]− tetrahedra including jumps by a small angle from the equilibrium position. | ||||||||
 |
|
|||||||
We report the DFT study of the vibrational spectroscopy properties of Mg(B3H8)2, a potential intermediate in the decomposition of Mg(BH4)2, as well as those of CB11H12− and CB9H10−, whose salts can exhibit high ionic conductivities. Because the inclusion of anharmonicity is key to the accurate description of the vibrational properties of BH species [D. Sethio, L. M. Lawson Daku, H. Hagemann. Int. J. Hydrogen Energy, 41 (2016) 6814], the calculations were performed both in the harmonic and in the anharmonic approximation. The IR and Raman spectra of Cs(CB11H12) and Na2(B10H10) have also been measured. The calculated and experimental spectra are in good agreement. A comparative analysis of the vibrational spectroscopy properties is made for B3H8− and Mg(B3H8)2, B12H122− and CB11H12−, and for B10H102− and CB9H10−. | ||||||||
|
 |
|||||||
Na2(B12H12)0.5(B10H10)0.5, a new solid-state sodium electrolyte is shown to offer high Na+ conductivity of 0.9 mS cm-1 at 20 °C, excellent thermal stability up to 300 °C, and a large electrochemical stability window of 3 V including stability towards sodium metal anodes, all essential prerequisites for a stable room-temperature 3 V all-solid-state sodium-ion battery. | ||||||||
|
||||||||
Alkaline or alkaline earth octahydrotriborate M(B3H8)x and dodecahydro-closo-dodecaborate MxB12H12 (M = Li, Na, Mg or Ca with x = 1 or 2) have recently attracted a lot of interest for hydrogen storage and solid electrolyte applications. Nevertheless, their syntheses are still a roadblock for large scale applications. In this paper we propose a novel approach for their syntheses starting from the cheapest borohydride NaBH4. The process involves first the solvothermal synthesis of tetrabutylammonium octahydrotriborate (C4H9)4NB3H8 (TBAB3H8) being the basis for the syntheses of the others boranes. Starting from TBAB3H8, we have synthesized pure and unsolvated NaB3H8 by salt metathesis reaction with sodium tetraphenylborate. Then, we have successfully obtained Na2B12H12 by solvothermal decomposition of NaB3H8. This approach has shown to be quantitative and reproducible, which could lead to the development of these boranes in real life applications. | ||||||||
 |
|
|||||||
This work deals with the synthesis and crystal structure study of new bismuth oxo-arsenates and their oxo-phosphates homologous: Bi6ZnO7(AsO4)2 vs Bi6ZnO7(PO4)2 and Bi3.667Cd3O4(AsO4)3 vs Bi3Cd4O4(PO4)3. Their crystal structures were solved using single crystal X-Ray Diffraction. These are two other examples of crystal structures built on ribbon-like polycations formed of the linkage of oxo-centered O(Bi,M)4 tetrahedra sharing edges and surrounded by isolated XO4 groups (X= As or P), where the O(Bi,M)4 units are derived from the fluorite topology structure. Dealing with Bi6ZnO7(PO4), its acentric space group was confirmed by preliminary second harmonic generation (SHG). The P/As substitution led to a centrosymmetric space group due to local reorientation of oxo-anions. This is strongly related to steric effects between AsO4 (d As-O= 1.6-1.7Å) and PO4 (d P-O= 1.4-1.5Å). Concerning Bi3.667Cd3O4(AsO4)3 and Bi3Cd4O4(PO4)3, they show a second example of the reorientation of the XO4 groups depending of the X chemical nature. Finally, we present an original topology of oxo-centered units obtained with Bi5KO5(AsO4). The photoluminescence properties of Bi5KO5(AsO4) and Bi6ZnO7(AsO4)2 were also investigated. The first one emits at room temperature in the reddish-orange range (single band peak at 615nm assigned to the Bi3+: 3P1→1S0 transition whereas the second exhibits a weak emission in the green range (peak at 530nm). Its intriguing temperature dependence is discussed in the paper. | ||||||||
|
 |
|||||||
Borohydrides have attained high interest in the past few years due to their high volumetric and gravimetric hydrogen content. Synthesis of di/trimetallic borohydride is a way to alter the thermodynamics of hydrogen release from borohydrides. Previously reported preparations of M(BH4)2 involved chloride containing species such as SrCl2. The presence of residual chloride (or other halide) ions in borohydrides may change their thermodynamic behavior and their decomposition pathway. Pure monometallic borohydrides are needed to study decomposition products without interference from halide impurities. They can also be used as precursors for synthesizing di/trimetallic borohydrides. In this paper we present a way to synthesize halide free alkaline earth metal (Sr, Ba) and europium borohydrides starting with the respective hydrides as precursors. Two novel high temperature polymorphs of Sr and Eu borohydrides and four polymorphs of Ba borohydride have been characterized by synchrotron X-ray powder diffraction, thermal analysis, and Raman and infrared spectroscopy and supported by periodic DFT calculations. The decomposition routes of these borohydrides have also been investigated. In the case of the decomposition of strontium and europium borohydrides, the metal borohydride hydride (M(BH4)H3, M = Sr, Eu) is observed and characterized. Periodic DFT calculations performed on room temperature Ba(BH4)2 revealed the presence of bidentate and tridentate borohydrides. | ||||||||

 |
|
|||||||
Two reactive hydride composite systems, Ca(BH4)2–NaNH2 and Mg(BH4)2–NaNH2, were systematically studied by in situ synchrotron radiation powder diffraction, in situ Fourier transform infrared spectroscopy, thermogravimetric analysis and differential scanning calorimetry coupled with mass spectrometry. Metathesis reactions between the amides and borohydrides take place in both systems between 100°C and 150°C yielding amorphous materials with the proposed composition M(BH4)(NH2). Simultaneously, a fraction of NaNH2 decomposes to Na3N and ammonia via a complex pathway. The main gas released under 300°C is ammonia for both systems, while significant amounts of hydrogen are released only above 350°C. | ||||||||
|
 |
|||||||
The characterization of boron-hydrogen compounds is an active research area which encompasses subjects as diverse as the chemistry and structures of closoboranes or the thermal decomposition mechanism of the borohydrides. Due to their high gravimetric hydrogen content, borohydrides are considered as potential hydrogen storage materials. Their thermal decompositions are multistep processes, for which the intermediate products are not easily identified. To help address this issue, we have extensively investigated the vibrational and NMR properties of 21 relevant Bm boron-hydrogen species (m = 1–12; n = 1–14; z = 0–2) within density functional theory. We could thus show that the B3LYP-D2 dispersion-corrected hybrid can be used in combination with the large cc-pVTZ basis set for the reliable prediction of the 11B and 1H NMR spectra of the boron-hydrogen species, and also for the reliable prediction of their IR and Raman spectra while taking into account the anharmonicity of their molecular vibrations. | ||||||||
 |
|
|||||||
SrAl2O4 doped with europium and dysprosium is a powerful and widely used afterglow material. Within this material strontium is found in two crystallographic different sites. Due to the similar ion radii and same charge, Eu2+-ions can occupy both sites, resulting in two different Eu2+-ions, one emitting in the blue and one in the green spectral range. The blue emission is thermally quenched at room temperature. In this paper we investigate the energy transfer between different Eu ions depending on the concentration and temperature using two different approaches: lifetime measurements and integrated intensity. We find an activation energy for the thermal quenching of the blue emission of 0.195 ± 0.023 eV and a critical radius for the energy transfer of 3.0 ± 0.5 nm. This results can help in designing better afterglow materials due to the fact that with energy transfer parts of the lost emission in the blue region at room temperature can be converted to the green site. | ||||||||
|
||||||||
The persistent phosphorescence and thermoluminescence of SrAl2O4:Eu2+:Dy3+ is reported for a variety of different excitation wavelengths and excitation temperatures, to provide new insights in the mechanism of the trapping and detrapping. These measurements reveal that the trapping is strongly dependent on the wavelength and temperature. First, with increasing loading temperature, the thermoluminescence peak shifts to lower temperatures which corresponds to a change of trap population. Secondly, the integrated thermoluminescent intensity increases with increasing loading temperature. All wavelength and temperature dependent experiments indicate that the loading of the traps is a thermally activated processes. Utilizing different wavelengths for loading, this effect can be enhanced or reduced. Furthermore excitation with UV-B-light reveals a tendency for detrapping the phosphor, reducing the resulting thermoluminescent intensity and changing the population of the traps. | ||||||||
|
 |
|||||||
The synthesis of 10 novel P-substituted dithienophosphole oxide compounds applying phenylcarbazole and indolocarbazole donors is presented. Based on photo-physical and theoretical investigations, the study reveals that the pyramidal geometry of the phosphorus allows for the synthesis of charge transfer materials by introducing strong exocyclic donor groups but suppresses intramolecular charge transfer below a certain donor strength threshold, which is an appealing structural feature for the design of donor–acceptor materials. The triplet energies of the phenylcarbazole based compounds are in the range of 2.49–2.65 eV, sufficiently high for potential applications as host materials in PhOLEDs. By contrast, the introduction of indolocarbazole, the weakest employed donor, yields materials exhibiting a significantly higher triplet energy of up to 2.87 eV and a remarkably low singlet–triplet splitting (0.18 eV). In addition an interesting example of an intramolecular electronic through-space interaction has been observed for the ortho-linked phenylcarbazole derivative. | ||||||||
 |
|
|||||||
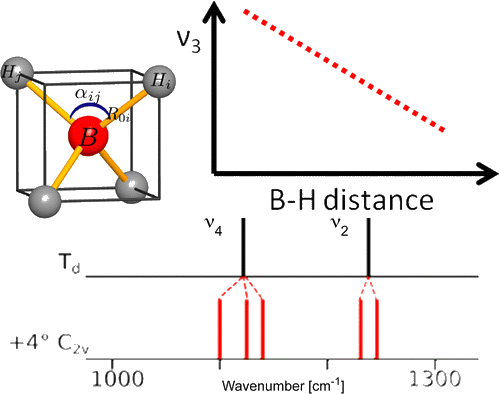
Among the different potential hydrogen storage materials, borohydrides have been largely investigated because of their high gravimetric and volumetric hydrogen content. In the analysis of borohydrides, vibrational spectroscopy plays an important role since it gives information on the local structure of the BH4– ion inside the solid. Here the GF method, developed by Wilson, is used in order to determine the local symmetry of BH4– in solid borohydrides starting from their vibrational spectra. Two different cases of deformations of BH4– are considered. In the first case, the effects of small angular variations on the vibrational spectra of borohydrides will be taken into account; starting from the splitting of the bands corresponding to the deformation modes, the angular deformations will be estimated. In the second one, the BH4– under chemical pressure (in different cubic alkali halides) is considered; in this case, the symmetry of the BH4– remains Td, while the bond lengths change according to the pressure experienced. Different practical examples will be illustrated. | ||||||||
|
 |
|||||||
can be formed during the thermal decomposition of metal borohydrides (M(BH4)x). Halogen ions such as fluoride or chloride can contribute to destabilize the ions. Hydride and fluoride mixed species like will be probable products after hydrogen release from mixed boro-hydride-fluoride or borohydride-borofluoride systems (, ). Various number of isomers are possible for (n = 2–11). DFT calculations were performed on isolated ions of all the possible isomers for (n = 0–3, 9–12), using B3LYP functionals and 6-31G(d,p) basis set. Relative stability, vibrational and NMR spectroscopy of these isomers are discussed and compared with available experimental data. | ||||||||
|
||||||||
A bimetallic dodecaborate LiNaB12H12 has been successfully synthesized for the first time, through a sintering process of LiBH4, NaBH4 and B10H14. LiNaB12H12 has a cubic Pa-3 space group symmetry at room temperature, and transforms into a high temperature phase with Fm-3m symmetry at 488 K, which is lower than that of Li2B12H12 and Na2B12H12. The ionic conductivity at 550 K reaches 0.79 S/cm, which is approximately 8 times higher than that of Na2B12H12 and 11 times higher than that of Li2B12H12. The Li/Na compositional and thus an induced positional disorder in LiNaB12H12 are suggested to be responsible for the reduced phase transition temperature and the improved super ionic conductivity compared to its monometallic counterparts. | ||||||||
|
||||||||
Persistent luminescence of SrAl2O4:Eu2+ has attracted considerable attention due to their high initial brightness, long-lasting time and excellent thermal stability. Here the influence of boric acid on the persistent luminescence and thermal oxidation resistance of SrAl2O4:Eu2+ was investigated in detail. Crystal structural analysis and scanning electron microscopy revealed that with the addition of boron, the unit cell volume decreased and the morphology of the particles became more irregular with sharp edges. Thermogravimetric analysis showed better thermal oxidation resistance accompanied by a change in oxygen vacancy concentration when boron acid is used. Photoluminescence spectra and afterglow decay curves confirm an improved afterglow performance for boron-added SrAl2O4:Eu2+. Thermoluminesence allowed monitoring the changes in the trap states due to the presence of B. Our results imply that the substantial improvement of afterglow performance and the thermal stability in SrAl2O4:Eu2+ can be attributed to the incorporation of boron into the aluminate network. | ||||||||
 |
|
|||||||
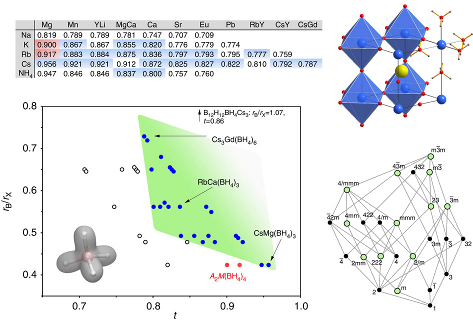
The structural phase transitions occurring in a series of perovskite-type complex hydrides based on the tetrahydroborate anion BH4- are investigated by means of in situ synchrotron x-ray powder diffraction, vibrational spectroscopy, thermal methods and ab initio calculations in the solid state. Structural dynamics of the BH4 anion are followed with quasi-elastic neutron scattering. We show that unexpected temperature-induced lattice instabilities in perovskite-type ACa(BH4)3 (A = K, Rb, Cs) have their origin in close hydridic di-hydrogen contacts. The rich lattice dynamics lead to coupling between internal B-H vibrations and phonons, resulting in distortions in the high-temperature polymorph that are identical in symmetry to well-known instabilities in oxide perovskites, generally condensing at lower temperatures. It is found that anion-substitution BH4- ↔ X- (X = Halide) can relax distortions in ACa(BH4)3 by eliminating coulomb repulsive H- • • • H- effects. The interesting nature of phase transition in ACa(BH4)3 enters an unexplored field of weak interactions in ceramic-like host lattices and is the principal motivation for this study. Close di-hydrogen contacts suggest new concepts to tailor crystal symmetries in complex hydride perovskites in the future. | ||||||||
|
||||||||
Single crystals of tetragonal RCuGa3 (R=La, Pr, Nd and Gd), with BaNiSn3 type structure (space group I4 mm), have been grown by high temperature solution growth technique using Ga as flux. Their magnetic properties were determined by heat capacity and the measurement of magnetization and electrical resistivity along [100] and [001] directions. Except LaCuGa3, the compounds order magnetically. PrCuGa3 undergoes a ferromagnetic transition with Curie temperature of 4.6 K. NdCuGa3 shows a bulk magnetic transition at 3.3 K. The data on GdCuGa3 indicate combined characteristics of spin glass and antiferromagnetic behavior at low temperatures. From the Schottky heat capacity data the crystal electric field level energy spectra have been determined. Further we have performed temperature dependent measurements of electron spin resonance (ESR) on GdCuGa3 between 11 K and room temperature. The ESR data indicate an enhancement of magnetic fluctuations associated with spin reorientation and both homogeneous and inhomogeneous thermal broadening of the linewidth. | ||||||||
|
 |
|||||||
The absorption spectra of Sm2+ doped in MFX (M=Sr, Ba; X=Cl, Br) crystals were studied within the range of 20,000–35,000 cm−1 as a function of temperature and host. The absorption bands observed were described with a simple model developed by Wood and Kaiser using group theory. The temperature and host dependence on the 7F0→5D3 Fano resonance lines were investigated. BaFCl:Sm2+ system showed a “normal” 7F0→5D3 transition at 4 K in spite of similar crystal structure and absorption profile with other MFX hosts. New Fano resonances were observed in the absorption spectra at higher energies (23,000–25,000 cm−1) for all MFX:Sm2+ systems at 4 K which persist up to room temperature. Preliminary energy level calculation showed that these resonance lines involve the interaction between higher excited 5LJ states of 4 f6configuration and 4 f55d1 configuration. | ||||||||
 |
|
|||||||
A rational molecular design strategy for carbazole–oxadiazole based bipolar host materials was developed to improve the device efficiency of blue phosphorescent organic light-emitting diodes (PHOLED). Steric effects of strategically placed methyl groups led to an increase of triplet energies (o-2MPCzPOXD: 2.66 eV and o-3MPCzPOXD: 2.73 eV versus the initial host material o-PczPOXD: 2.62 eV) while less pronouncedly affecting singlet energies and, therefore, retaining low driving voltages, high power efficiencies and remarkably low efficiency roll-offs in PHOLEDs. The maximum quantum efficiencies (EQE) for blue devices (FIrpic) were significantly raised for o-2MPCzPOXD (13.6%) and o-3MPCzPOXD (11.5%) versus o-PCzPOXD (9.0%) although yielding comparable values for green devices (Ir(ppy)3; 12.9% and 15.4% versus 13.2%). Supported by theoretical calculations a structure–property relationship was established from photo-physical properties, PHOLED performance measurements and structural characterization from single crystal data. | ||||||||
|
 |
|||||||
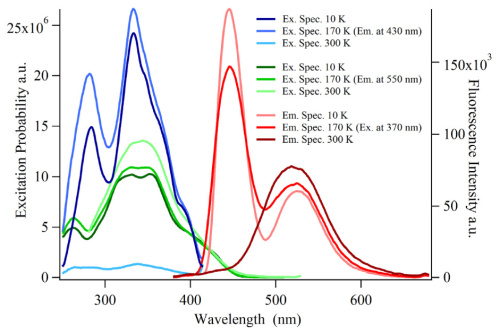
Europium doped crystalline Ba7F12Cl2 phosphors have been prepared at temperatures between 650 and 900 °C using alkali chloride fluxes, yielding both disordered (with the incorporation of small amounts of Na) and ordered crystal modifications. The white emission spectrum excited in the near UV consists roughly of two broad emission bands at ca 450 and 590 nm, as well as weak sharp Eu2+ 4f-4f emission bands around 360 nm. The incorporation of Eu2+ is further studied using EPR spectroscopy on single crystals, and reveals a significant zero field splitting. The emission spectrum can be significantly tuned by varying the excitation wavelength between 300 and 390 nm. Fine tuning may also be achieved by chemical substitutions to form Ba7-xMyF12Cl2-zBrz (M = Na, Ca,Eu). Quantitative measurements of the light produced using commercial near UV LEDs show that the color temperature ranges between 4000 and 9700 K with CIE chromaticity coordinates close to the ideal values of x=y=0.333. The best color rendering index (CRI) found was 0.83, and the highest light to light conversion yield was 171 lumen/W. These results show that the title compound is a very promising candidate for white light generation using near UV LED excitation. | ||||||||
|
||||||||
Borohydrides are actively considered as potential hydrogen storage materials. In this context fundamental understanding of breaking and forming B-H bond is essential. Isotope exchange reactions allow isolating some parts of this reaction without introducing major structural or chemical changes. Experiments were performed on Ca(BH4)2and Ca(BD4)2 as a function of temperature and pressure. A complete exchange can be realized in about 9h at 200 °C using a deuterium pressure of 20 bar. The activation energy, estimated using first order kinetics, for the forward reaction (Ca(BH4)2 → Ca(BD4)2) was found to be 82.1 ± 2.7 kJ/mol (P = 35 bar) and the one for the backward reaction (Ca(BD4)2 → Ca(BH4)2) was found to be 98.5 ± 8.3 kJ/mol (P = 35 bar). Pressure dependent study shows that the reaction rate increases with increasing pressure up to 35 bar. This behavior is consistent with first adsorption step prior to diffusion into the solid and isotope exchange according to the scheme described below. | ||||||||
 |
|
|||||||
To celebrate the International Year of Crystallography among the general public, a consortium of chemists, physicists and crystallographers of the University of Geneva organised in Spring 2014 an incentive crystal growth contest for Geneva scholars aged 4 to 19. Starting from a kit containing a salt and user instructions, classes had to prepare a crystal that met specific criteria according to their category of age. The composition of the salt – potassium dihydrogen phosphate (KDP) – was only disclosed to the participants during the final Awards Ceremony. This contest positively exceeded our expectations with almost 100 participating classes (ca. 1800 participants) and 54 specimens received over all categories. | ||||||||
|
 |
|||||||
Perovskite materials host an incredible variety of functionalities. Although the lightest element, hydrogen, is rarely encountered in oxide perovskite lattices, it was recently observed as the hydride anion H−, substituting for the oxide anion in BaTiO3. Here we present a series of 30 new complex hydride perovskite-type materials, based on the non-spherical tetrahydroborate anion BH4− and new synthesis protocols involving rare-earth elements. Photophysical, electronic and hydrogen storage properties are discussed, along with counterintuitive trends in structural behaviour. The electronic structure is investigated theoretically with density functional theory solid-state calculations. BH4-specific anion dynamics are introduced to perovskites, mediating mechanisms that freeze lattice instabilities and generate supercells of up to 16 × the unit cell volume in AB(BH4)3. In this view, homopolar hydridic di-hydrogen contacts arise as a potential tool with which to tailor crystal symmetries, thus merging concepts of molecular chemistry with ceramic-like host lattices. Furthermore, anion mixing BH4−←X− (X−=Cl−, Br−, I−) provides a link to the known ABX3 halides. | ||||||||
|
||||||||
Hydrolysis of metal borohydrides in the presence of CO2 has not been studied so far, although carbon dioxide contained in air is known to accelerate hydrogen generation. KBH4 hydrolysis promoted by CO2 gas put through an aqueous solution was studied by time-resolved ATR-FTIR spectroscopy, showing a transformation of BH4− into B4O5(OH)42−, and a drastically accelerated hydrogen production which can be completed within minutes. This process can be used to produce hydrogen on-board from exhaust gases (CO2 and H2O). We found a new intermediate, K9[B4O5(OH)4]3(CO3)(BH4)·7H2O, forming upon hydrolysis on air via a slow adsorption of the atmospheric CO2. The same intermediate can be crystallized from partly hydrolyzed solutions of KBH4 + CO2, but not from the fully reacted sample saturated with CO2. This phase was studied by single-crystal and powder X-ray diffraction, DSC, TGA, Raman, IR and elemental analysis, all data are fully consistent with the presence of the three different anions and of the crystallized water molecules. Its crystal structure is hexagonal, space group P-62c, with lattice parameters a = 11.2551(4), c = 17.1508(8) Å. Formation of the intermediate produces 16 mol of H2 per mole of adsorbed CO2 and thus is very efficient both gravimetrically and volumetrically. It allows also for an elimination of carbon dioxide from exhaust gases. | ||||||||
|
||||||||
In analogy to the synthesis of polycrystalline M2NaIO6 (M = Ca, Sr, Ba) by precipitation in water at 90 °C, the title compound was first prepared as a metastable compound. The stable modification of Pb2NaIO6 was obtained by a heat treatment to 400 °C followed by cooling to room temperature. The crystal structure was refined from powder diffraction data [space group P21/c (14), a = 5.9040(2), b = 5.7526(2), c = 10.1104(3) Å, β = 125.341(1)°]. On heating, at ca. 125 °C, a phase transition to a cubic high temperature modification was observed. The crystal structure was refined from XRD data measured at 200 °C [space group Fm3m (225), a = 8.2678(1) Å]. Depending on the precipitation temperature between 90 °C and 0 °C, several metastable modifications were obtained, which can be distinguished by significantly different lattice parameters. The XRD pattern of a powder precipitated at room temperature is pseudocubic. The crystal structure was refined at room temperature in P21/c with a = 5.8201(4), b = 5.8473(4), c = 10.0798(5) Å, β = 125.074(3)°. This modification behaves almost as a cubic lattice on heating as found from XRD and DSC measurements. | ||||||||
 |
|
|||||||
The compound Ba5I2O12 was synthesized by heating a precipitate of dissolved Ba(OH)2·8H2O and H5IO6. Rb2O was added to increase the crystallite size. The crystal structure was determined from conventional laboratory X-ray diffraction data by using a real-space structure solution approach followed by a Rietveld refinement. No constraints on positions were used. The structure analysis gave an orthorhombic symmetry with a = 19.7474(2) Å, b = 5.9006(1) Å and c = 10.5773(1) Å. The final RBragg value in space group Pnma (62) was 1.0 %. The structure can be described by layers of a metal and iodine arrangement forming almost pentagonal holes. Raman measurements were correlated with the two IO6 octahedra. Two further barium periodate patterns were observed and indexed. | ||||||||
|
 |
|||||||
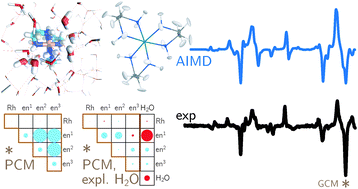
Backscattered Raman optical activity (ROA) spectra are measured for Δ- and Λ-tris-(ethylenediamine)rhodium(III) chloride in aqueous solution. In addition, the spectra of the four possible conformers in the Λ configuration are investigated by ab initio calculations. The Λ(δδδ) conformer is in best agreement with experimental spectra and examined in more details. The two most stable conformers according to the calculations are not compatible with the experimental ROA spectrum. Insights into the origin of observed band intensities are obtained by means of group coupling matrices. The influence of the first solvation shell is explored via an ab initio molecular dynamics simulation. Taking explicit solvent molecules into account further improves the agreement between calculation and experiment. Analysis of selected normal modes using group coupling matrices shows that solvent molecules lead to normal mode rotation and thus contribute to the ROA intensity, whereas the contribution of the Rh can be neglected. | ||||||||
|
||||||||
Fluorine-substituted CaTiO3:Pr phosphors were prepared by a solid-state reaction. Rietveld refinements of powder X-ray diffraction patterns revealed that increasing fluorine-substitution leads to the gradual shrinkage of the unit-cell. Enhanced afterglow intensities were observed with fluorine-substitution. Furthermore, the effect of annealing atmosphere was investigated by thermochemical treatment in different atmospheres (Ar, air and NH3). UV-Vis diffuse reflectance spectra and photoluminescence excitation spectra revealed that Pr4+ in the pristine CaTi(O,F)3:Pr phosphor was partially reduced to Pr3+ under NH3 flow leading to an intensity improvement of ca. 450% compared to CaTiO3:Pr. The substantial improvement of afterglow intensity by fluorine substitution and annealing in NH3 is considered to be connected with the generation of oxygen vacancies and the partial reduction of Pr4+ to Pr3+. | ||||||||
 |
|
|||||||
Inorganic compounds with BH4- ions are the subject of many recent investigations in the context of potential hydrogen storage materials. In this work, Attenuated Total Reflectance Fourier Transform Infrared (ATR-FTIR) spectra of a series of reference and research compounds (including deuterated samples) are collected and made available to the research community. | ||||||||
|
||||||||
We report on a comprehensive study of the magnetization, resistivity and heat capacity on the single crystals of Ce2RhGa12 synthesized using Ga flux. Single crystal X-ray diffraction data confirm the tetragonal Pb/nbm structure of Ce2RhGa12, which is isostructural to Ce2PdGa12. Ce2RhGa12 orders antiferromagnetically at TN = 3.5 K and exhibits anisotropic magnetic behavior, inferred from the magnetization and resistivity data, taken along the two principal crystallographic directions of the crystal, viz., along [100] and [001]. The anisotropic magnetic response of Ce2RhGa12 establishes [001] as the easy axis of magnetization, and a weak meta-magnetic transition is also observed in the magnetic isotherm at 2K along the same axis. A sharp peak in specific heat signals the bulk antiferromagnetic transition at TN = 3.5 K, which shifts to lower temperatures in low applied fields. The electrical resistivity along the two directions shows metallic behavior from 300K down to 1.8K and establishes Ce2RhGa12 as a normal, trivalent cerium compound. | ||||||||
|
 |
|||||||
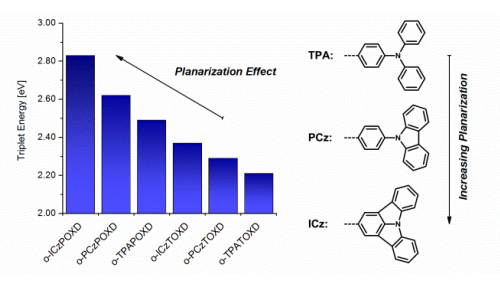
A series of 6 novel triarylamine-containing oxadiazole compounds (o-PCzPOXD, o-ICzPOXD, o-TPATOXD, o-PCzTOXD, o-ICzTOXD, o-CzTOXD) have been designed, synthesized and characterized concerning applications as host materials in PHOLED devices. To further improve the ortho-linkage concept, the impact of incorporating planarized electron-donating triarylamine (TAA) structures on intramolecular charge transfer was examined. The effect was evaluated for two series of electron-accepting oxadiazole scaffolds, realizing ortho-linkage on the benzene (POXD) and the thiophene (TOXD) core. Thermal analysis shows increased glass-transition temperatures for planarized structures indicating an improved morphological stability. A higher degree of planarization also results in significantly increased singlet and triplet energy values, revealing the impact on the intramolecular charge transfer. Employing the developed materials, red (o-TPATOXD: CEmax: 28.8 cd A-1, EQEmax: 16.9%), green (o-PCzPOXD: CEmax: 62.9 cd A-1, EQEmax: 17.1%) and blue (o-PCzPOXD: CEmax: 29.8 cd A-1, EQEmax: 13.4%) devices were achieved showing remarkably low efficiency roll-off for planarized donors. Hence, this is the first report of efficient blue devices for this specific class of host materials. It is proposed that the results correlate with an increasing ortho-linkage effect and decreasing donor strength of the TAA moiety by planarization and, thus, tackling one of the major challenges in PHOLED research: improving both triplet energy and compound stability. | ||||||||
 |
|
|||||||
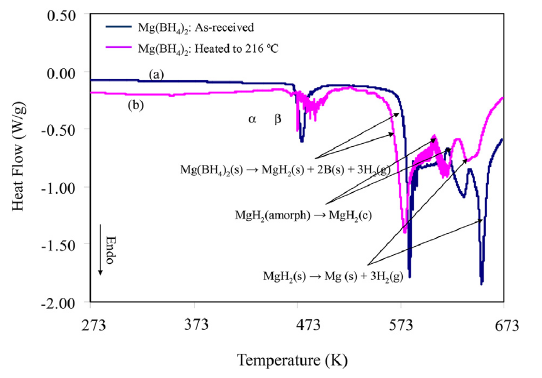
The vapor pressure and molecular weight of effusing vapors of α, β, and amorphous Mg(BH4)2 were determined by Torsion-effusion gravimetric method, under dynamic vacuum. A Cahn balance in the system yielded the rate of the weight loss. Molecular weights measured revealed if the effusion was congruent or there was disproportionation. The vaporization behavior of crystalline Mg(BH4)2, was measured up to 533 K at pressures of ∼10−5 torr. It was found that Mg(BH4)2 disproportionates to form predominantly H2 gas (∼95%) with a small amount of Mg(BH4)2 (∼5%) in the gas phase. The combined average molecular weight measured is 4.16 g/mol. The equations for vapor pressures for crystalline Mg(BH4)2 are given by: log PTotal(bar) = 9.2303 − 7286.2/T, log PMg(BH4)2 (bar) = 8.2515 - 7286.2 / T , and log PH2 (bar) = 9.1821 - 7286.2 / T. The partial pressures of the gaseous species were determined as PMg2(4BH)(g)/PT=0.105 and PH2(g)/PT=0.895. Enthalpies of vaporization for the effusing gases were calculated to be ΔH = +558.0 kJ/mol H2 and ΔH = +135 kJ/mol Mg(BH4)2. The standard Gibbs free energy changes, ΔG°(kJ/mol), for the complete decomposition reaction (Mg(BH4)2(s) → Mg(s) + 2B(s) + 4H2(g)), sublimation reaction (Mg(BH4)2(s) → Mg(BH4)2(g)) and the disproportionation reaction for Mg(BH4)2 are reported in this paper. The decomposition pathway of amorphous Mg(BH4)2 was also carried out between 388.2 K and 712.8 K showing multistep decomposition of a-Mg(BH4)2 Different reaction products were obtained depending on the method used in the vaporization experiment. The behavior of the amorphous Mg(BH4)2(s) is very different from those for the two crystalline phases (α and β). The vapor pressure behavior and thermodynamics of vaporization of different phases of Mg(BH4)2 are presented. | ||||||||
|
 |
|||||||
Hydrogen-fluorine exchange in the NaBH4–NaBF4 system is investigated with a range of experimental methods combined with DFT calculations and a possible mechanism for the reactions is proposed. Fluorine substitution is observed by in-situ synchrotron radiation powder X-ray diffraction (SR-PXD) as a new Rock salt type compound with idealized composition NaBF2H2 in the temperature range T = 200 to 215 °C. Combined use of solid-state 19F MAS NMR, FT-IR and DFT calculations supports the formation of a BF2H2− complex ion, reproducing the observation of a 19F chemical shift at 144.2 ppm, which is different from that of NaBF4 at 159.2 ppm, along with the new absorption bands observed in the IR spectra. After further heating, the fluorine substituted compound becomes X-ray amorphous and decomposes to NaF at ~310 ºC. This work shows that fluorine-substituted borohydrides tend to decompose to more stable compounds, e.g. NaF, BF3 or amorphous products such as closo-boranes, e.g. Na2B12H12. The NaBH4-NaBF4 composite decomposes at lower temperatures (300 °C) compared to NaBH4 (476 °C), as observed by thermogravimetric analysis. NaBH4-NaBF4 (1:0.5) preserves 30 % of the hydrogen storage capacity after three hydrogen release and uptake cycles compared to 8 % for NaBH4 measured by the Sievert’s method under identical conditions, but more than 50 % using prolonged hydrogen absorption time. The reversible hydrogen storage capacity tends to decrease possibly due to the formation of NaF and Na2B12H12. On the other hand, the additive sodium fluoride appears to facilitate hydrogen uptake, prevent foaming, phase segregation and loss of material from the sample container for samples of NaBH4-NaF. | ||||||||
 |
|
|||||||
The crystal structures of the M2NaIO6 series (M = Ca, Sr, Ba), prepared at 650 °C by ceramic methods, were determined from conventional laboratory X-ray powder diffraction data. Synthesis and crystal growth were made by oxidizing I– with O2(air) to I7+ followed by crystal growth in the presence of NaF as mineralizator, or by the reaction of the alkali-metal periodate with the alkaline-earth metal hydroxide. All three compounds are insoluble and stable in water. The barium compound crystallizes in the cubic space group Fm3m (no. 225) with lattice parameters of a = 8.3384(1) Å, whereas the strontium and calcium compounds crystallize in the monoclinic space group P21/c (no. 14) with a = 5.7600(1) Å, b = 5.7759(1) Å, c = 9.9742(1) Å, β = 125.362(1)° and a = 5.5376(1) Å, b = 5.7911(1) Å, c = 9.6055(1) Å, β = 124.300(1)°, respectively. The crystal structure consists of either symmetric (for Ba) or distorted (for Sr and Ca) perovskite superstructures. Ba2NaIO6 contains the first perfectly octahedral [IO6]5– unit reported. The compounds of the ortho-periodates are stable up to 800 °C. Spectroscopic measurements as well as DFT calculations show a reasonable agreement between calculated and observed IR- and Raman-active vibrations. | ||||||||
|
 |
|||||||
The emission lifetime of Sm2+ ions doped in MFX (M=Ba, Sr; X=Br, I) crystals was investigated as a function of pressure and temperature. The decay of the 5DJ(J=0,1,2) levels showed single exponential relaxation. The analysis of these experiments yielded the position of the lowest 4f55d1 state as well as non-radiative rate constants. These values were compared with those for Sm2+ doped in other matlockite host crystals. The single exponential decrease of the 5D0,1 lifetime as a function of pressure was described considering the increased radiative decay rates of these 5D0,1 levels through electronic mixing between the 4f55d1 and 5DJstates. | ||||||||
|
||||||||
The melting behavior, the solubility, and the influence of hydrogen bonds were analyzed for a series of single- and double-tailed surfactant alcohols. Various effects such as the presence of free amides or the intermolecular spacing were found to be important factors for increasing or decreasing the melting temperature of a surfactant. Furthermore, we present a model for the packing of diamido-lipids and study the temperature-dependence of the IR signals. | ||||||||
 |
|
|||||||
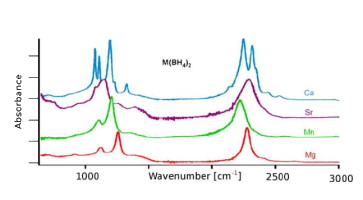
A comparison of the vibrational spectra of many inorganic borohydrides allows us to distinguish compounds with isolated BH4- ions and compounds containing complex ions such as Sc(BH4)4-. The characteristic spectral features of both types of compounds are identified, showing that the B–H bonding is quite different in both cases. A detailed analysis of the vibrations of the isolated BH4- ions provides new information about their local structure. Angular deformations of individual borohydride ion are analyzed quantitatively. It appears that the compounds containing isolated BH4- ions belong to those with the most electropositive cations and the highest decomposition temperature, while the complex borohydrides show significantly lower decomposition temperatures and possible diborane formation. | ||||||||
|
 |
|||||||
The phosphor CaTiO3:Pr3+ was synthesized via a solid-state reaction in combination with a subsequent annealing under flowing NH3. Comparatively large off-center displacements of Ti in the TiO6 octahedra were confirmed for as-synthesized CaTiO3:Pr3 by XANES. Raman spectroscopy showed that the local crystal structure becomes highly symmetric when the powders are ammonolyzed at 400 °C. Rietveld refinement of powder X-ray diffraction data revealed that the samples ammonolyzed at 400 °C have the smallest lattice strain and at the same time the largest average Ti-O-Ti angles were obtained. The samples ammonolyzed at 400 °C also showed the smallest mass loss during the thermal re-oxidation in thermogravimetric analysis (TGA). Enhanced photolumincescence brightness and an improved decay curve as well as the highest reflectance were obtained for the samples ammonolyzed at 400 °C. The improved photoluminescence and afterglow by NH3 treatment are explained as a result of the reduced concentration of oxygen excesses with simultaneous relaxation of the lattice strain. | ||||||||
 |
|
|||||||
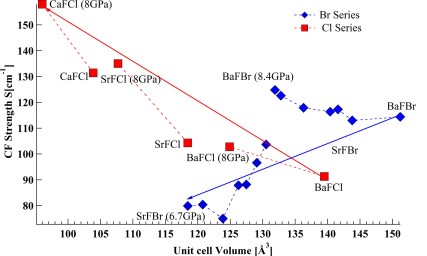
The emission spectra of Sm2+ doped in BaFBr and SrFBr hosts were measured at 10 K from ambient pressure to 8 GPa. The crystal field energy levels determined from the emission spectra were used to extract the free ion parameters (Fk and ζ ) and crystal field parameters (Bqk). The variation of Fk and ζ as a function of pressure was studied systematically and was discussed in relation to the central field and symmetry restricted covalency models. The change of the spin orbit coupling parameter (ζ) with pressure for SrFBr:Sm2+ showed very different behavior than in other matlockite hosts. Moreover the variation of Bqk under pressure was studied. The pressure dependence of the Bqk was described quantitatively using the Superposition Model (SM) with the help of structural parameters as a function of pressure, obtained from periodic DFT calculations. The validity of the SM was tested for Sm2+ in BaFBr and SrFBr. It is shown that this model does not apply to SrFBr, in contrast to other matlockite host materials. | ||||||||
|
 |
|||||||
Four novel bimetallic borohydrides have been discovered, K2M(BH4)4 (M = Mg or Mn), K3Mg(BH4)5, and KMn(BH4)3, and are carefully investigated structurally as well as regarding their decomposition reaction mechanism by means of in situ synchrotron radiation powder X-ray diffraction (SR-PXD), vibrational spectroscopies (Raman and IR), thermal analysis (TGA and DTA), and ab initio density functional theory (DFT) calculations. Mechano-chemical synthesis (ball-milling) using the reactants KBH4, α-Mg(BH4)2, and α-Mn(BH4)2 ensures chlorine-free reaction products. A detailed structural analysis reveals significant similarities as well as surprising differences among the two isomorphs K2M(BH4)4, most importantly concerning the extent to which the complex anion [M(BH4)4]2– is isolated in the structure. Anisotropic thermal expansion and an increase in symmetry at high temperatures in K3Mg(BH4)5 is ascribed to the motion of BH4 groups inducing hydrogen repulsive effects, and the dynamics of K3Mg(BH4)5 are investigated. Decomposition in the manganese system proceeds via the formation of KMn(BH4)3, the first perovkite type borohydride reported to date. | ||||||||
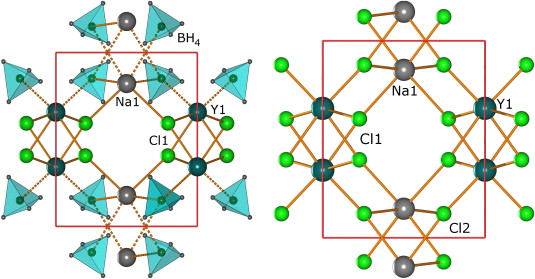 |
|
|||||||
Hydrogen production from waste feedstocks using supercritical water gasification (SCWG) is a promising approach towards cleaner fuel production and a solution for hard to treat wastes. In this study, the catalytic co-gasification of starch and catechol as models of carbohydrates and phenol compounds was investigated in a batch reactor at 28 MPa, 400–500 °C, from 10 to 30 min. The effects of reaction conditions, and the addition of calcium oxide (CaO) as a carbon dioxide (CO2) sorbent and TiO2 as catalyst on the gas yields and product distribution were investigated. Employing TiO2 as a catalyst alone had no significant effect on the H2 yield but when combined with CaO increased the hydrogen yield by 35% and promoted higher total organic carbon (TOC) reduction efficiencies. The process liquid effluent was characterized using GC–MS, with the results showing that the major non-polar components were phenol, substituted phenols, and cresols. An overall reaction scheme is provided. | ||||||||
|
 |
|||||||
Red emitting CaTiO3:Pr phosphors with a nominal composition of Ca0.998+xPr0.002TiO3+δ (0.02≤x≤0.04) were prepared by solid state reactions with different thermal post treatments and characterized by X-ray diffraction, transmission electron microscopy and photoluminescence. The Ca excess exhibited complete solubility up to 4% in the samples treated at 1400 °C but segregation in the form of Ruddlesden-Popper phases (Ca3Ti2O7 - Ca4Ti3O10) was observed in samples prepared at 1500 °C. The increase in temperature for stoichiometric samples showed a monotonic increase of decay time due to the reduction of non-radiative recombination defects. It was found that the Ca excess favored the formation of oxygen vacancies which are known to act as trap. In the samples treated at 1400 °C, 3% of Ca excess showed to be the best concentration to increase the decay time of persistent luminescence. For the samples treated at 1500 °C, the segregation of Ruddlesden-Popper phases left a constant amount of Ca soluble in all the CaTiO3 samples. This constant concentration of Ca caused the same density of defects and, consequently, the same decay time in all samples. | ||||||||
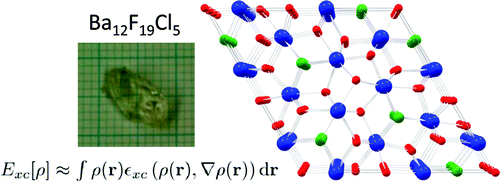 |
|
|||||||
The crystal chemistry of the barium fluoride chloride system is studied both experimentally and theoretically. Different synthetic approaches yield nanocrystalline materials as well as large single crystals. The crystalline phases identified so far are BaFCl, Ba12F19Cl5 and Ba7F12Cl2 (in two modifications) and compared with analogous compounds. It is demonstrated that the compound Ba2F3Cl reported by Fessenden and Lewin 50 years ago corresponds to Ba7F12Cl2. The phase diagram of the BaCl2 – BaF2 system is reinvestigated for fluoride mole fractions between 0.5 and 1. The peritectic formation of Ba12F19Cl5 is observed. Periodic DFT calculations are performed for all structures in this system, including a hypothetical structure for Ba2F3Cl, based on the experimental structure of Ba2H3Cl. The energy of formation of the different barium fluoride chloride compounds from BaCl2 and BaF2 (normalized for one barium atom per formula unit), as calculated by DFT at 0K, is within only about ± 15 kJ/mol. Comparison with recent experimental results on calcium and strontium hydride chloride (bromide) compounds, suggest the possibility of a mutual exclusion between the M2X3Y and M7X12Y2 (M = Ca, Sr, Ba, Pb, X = H, F, Y = Cl,Br) structures. The single crystal structure of PbFBr is also reported. | ||||||||
|
 |
|||||||
To study the reorientational motion of BH4 groups in β and γ phases of Mg(BH4)2 and in α and β phases of Ca(BH4)2, we have performed nuclear magnetic resonance (NMR) measurements of the 1H and 11B spin–lattice relaxation rates in these compounds over wide ranges of temperature and resonance frequency. It is found that at low temperatures the reorientational motion in β phases of Mg(BH4)2 and Ca(BH4)2 is considerably faster than in other studied phases of these alkaline-earth borohydrides. The behavior of the measured spin–lattice relaxation rates in both β phases can be satisfactorily described in terms of a Gaussian distribution of activation energies Ea with the average Ea values of 138 meV for β-Mg(BH4)2and 116 meV for β-Ca(BH4)2. The α phase of Ca(BH4)2 is characterized by the activation energy of 286 ± 7 meV. For the novel porous γ phase of Mg(BH4)2, the main reorientational process responsible for the observed spin–lattice relaxation rate maximum can be described by the activation energy of 276 ± 5 meV. The barriers for reorientational motion in different phases of alkaline-earth borohydrides are discussed on the basis of changes in the local environment of BH4 groups. | ||||||||
|
||||||||
Single crystals of Ce2RhGa12 have been synthesized using Ga flux and their structure was determined by single-crystal X-ray diffraction. Ce2RhGa12 crystallizes in the tetragonal space group P4/nbm (No. 125), and is isostructural to Ce2PdGa12, with Z = 2 and lattice parameters a = 6.0405 Å and c = 15.706 Å. Data were collected at the Swiss Norwegian Beam Line at the European Synchrotron Facility, Grenoble, France. Laue diffraction was carried out to confirm the quality of the single crystal and showed well-defined spots and tetragonal symmetry. | ||||||||
 |
|
|||||||
The title compound, an achiral flexible molecule containing a 1,2,3-triazole structure as the acceptor subunit, crystallizes as a single enantiomorph in the space group P212121. The material exhibits nonlinear optical properties and is capable of second harmonic generation. Thus, the developed molecular scaffold represents an interesting novel type of NLO chromophore. | ||||||||
|
 |
|||||||
Highly occupied: A highly porous form of Mg(BH4)2 (see picture; Mg green, BH4 blue, unit cells shown in red) reversibly absorbs H2, N2, and CH2Cl2. At high pressures, this material transforms into an interpenetrated framework that has 79 % higher density than the other polymorphs. Mg(BH4)2 can act as a coordination polymer that has many similarities to metal–organic frameworks. | ||||||||

 |
|
|||||||
Barium calcium magnesium fluoride (Ba2(BaxCa1-x)Mg4F14, x=0.19-0.26) has been synthesized at 850 °C from precursors prepared by the solution precipitation method. Single crystals with composition of Ba2.200(2)Ca0.800(2)Mg4F14were obtained after prolonged heating. Lattice parameters from single crystal data are a = 12.4203(8) and c = 7.4365(5) Å [tetragonal, space group P42/mnm (No. 136)]. They increase with increasing barium concentration within a given stability window. The structure is built of a network of MgF6 octahedra forming a pyrochlore related channel system and isolated fluorine ions. Within the channels, heavy alkaline earth ions are located. The wide channel is filled with off-center positioned barium ions. The channel with a narrow cross section hosts both ions, Ca2+and Ba2+. The structure is isotypic with Pb3Nb4O12F2 but has a different coordination around Ba/Ca and Pb, respectively. Doped with ∼1% Eu(II), the compound shows intense blue luminescence under UV activation. | ||||||||
|
||||||||
A solid solution of magnesium and manganese borohydrides was studied by in situ synchrotron radiation X-ray powder diffraction and infrared spectroscopy. A combination of thermogravimetry, mass and infrared spectroscopy, and atomic emission spectroscopy were applied to clarify the thermal gas desorption of pure Mn(BH4)2 and a solid solution of composition Mg0.5Mn0.5(BH4)2. MgxMn(1−x)(BH4)2 (x = 0–0.8) conserves the trigonal structure of Mn(BH4)2 at room temperature. Manganese is dissolved in the hexagonal structure of α-Mg(BH4)2, with the upper solubility limit not exceeding 10 mol.% at room temperature. There exists a two-phase region of trigonal and hexagonal borohydrides within the compositional rangex = 0.8–0.9 at room temperature. Infrared spectra show splitting of various vibrational modes, indicating the presence of two cations in the trigonal MgxMn(1−x)(BH4)2 solid solutions, as well as the appearance of a second phase, hexagonal α-Mg(BH4)2, at higher magnesium contents. All vibrational frequencies are shifted to higher values with increasing magnesium content. The decomposition temperature of the trigonal MgxMn(1−x)(BH4)2 (x = 0–0.8) does not vary significantly as a function of the magnesium content (433–453 K). The desorbed gas contains mostly hydrogen and 3–7.5 mol.% diborane B2H6, as determined from analyses of the Mn(BH4)2 and Mg0.5Mn0.5(BH4)2 samples. An eutectic relation between α-Mg(BH4)2 and LiBH4 is observed. The solid solution MgxMn(1−x)(BH4)2 is a promising material for hydrogen storage as it decomposes at a similar temperature to Mn(BH4)2, i.e. at a much lower temperature than pure Mg(BH4)2 without significantly losing hydrogen weight capacity thanks to substitution of Mn by Mg up to 80 mol.%. The questions of diborane release and reversibility remain to be addressed. | ||||||||
|
 |
|||||||
The synthesis of a novel alkali-metal aluminium borohydride NaAl(BH4)xCl4−x from NaBH4 and AlCl3 using a solid state metathesis reaction is described. Structure determination was carried out using synchrotron powder diffraction data and vibrational spectroscopy. An orthorhombic structure (space group Pmn21) is formed which contains Na+ cations and complex [Al(BH4,Cl)4]−anions. Due to the high chlorine content (1 ≤ x ≤ 1.43) the hydrogen density of the borohydride is only between 2.3 and 3.5 wt.% H2 in contrast to the expected 14.6 wt.% for chlorine free NaAl(BH4)4. The decomposition of NaAl(BH4)xCl4−x is observed in the target range for desorption at about 90 °C by differential scanning calorimetry (DSC), in situ Raman spectroscopy and synchrotron powder X-ray diffraction. Thermogravimetric analysis (TG) shows extensive mass loss indicating the loss of H2 and B2H6 at about 90 °C followed by extensive weight loss in the form of chloride evaporation. | ||||||||
|
||||||||
YMn2 forms either interstitial YMn2Hx hydrides for x ≤ 4.5 or a complex YMn2H6 hydride when submitted to high hydrogen pressure. These compounds have been studied by inelastic neutron scattering (INS) in order to clarify the different modes of H vibration. The INS spectra of YMn2Hx hydrides are strongly dependent on the H content. YMn2H6 and YMn2D6 show broad bands, also observed by Raman and IR spectroscopy, assigned to H–Mn–H (or D) and Mn–H bending and stretching modes. Both ErMn2D6 and ErMn1.8Fe0.2D6 show, in addition to the H vibration mode, an intense band at 215 cm−1 which has been attributed to a magnetic excitation of Er3+ in view of its momentum transfer dependence. | ||||||||
|
||||||||
The structural and vibrational properties of the isostructural compounds Ca2FeH6 and Sr2RuH6 are determined by periodic DFT calculations and compared with their previously published experimental crystal structures as well as new experimental vibrational data. The analysis of the vibrational data is extended to the whole series of alkaline-earth iron and ruthenium hydrides A2TH6 (A = Mg,Ca,Sr; T = Fe, Ru) in order to identify correlations between selected frequencies and the T-H bond length. The bulk moduli of Ca2FeH6 and Sr2RuH6 have also been determined within DFT. Their calculated values prove to compare well with the experimental values reported for Mg2FeH6 and several other compounds of this structure. | ||||||||
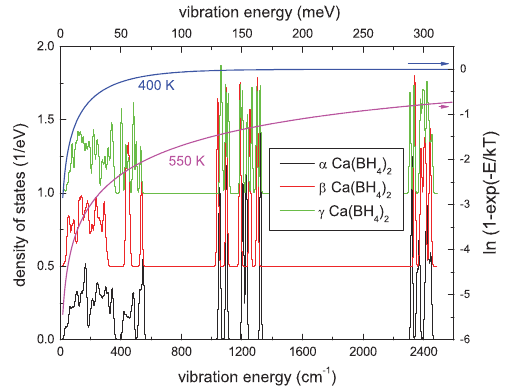 |
|
|||||||
The high energy of hydrogen vibrations in solids is the origin of their strong impact on thermodynamic properties. The peculiar structure of complex hydrides amplifies this impact. We shed light on the vibrational properties of three allotropes of Ca(BH4)2 using density-functional theory calculations, infrared spectroscopy, and inelastic neutron scattering. We show that the vibrational properties of Ca(BH4)2 depend on the specific phase and are hitherto the origin of their differences in stability. | ||||||||
|