

« Back to all publications
Download this list in a RIS file or a BIB file or a PDF file
|
||||||||
Using photoinduced bimolecular electron transfer reactions as example we demonstrate how diffusion controlled bimolecular chemical reactions can be studied in a model-free manner by quantitatively combining different ultrafast spectroscopical tools. | ||||||||




|
 |
|||||||
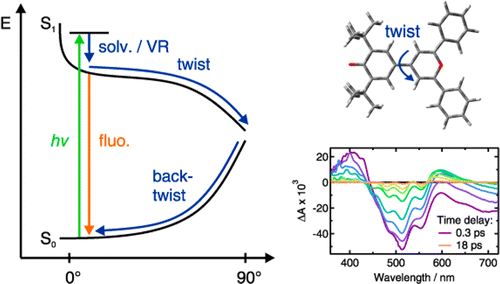
The excited-state dynamics of two donorâacceptor biaryls that differ by the strength of the acceptor, a pyridinium or a pyrylium moiety, have been investigated using a combination of steady-state solvatochromic absorption, ultrafast fluorescence, as well as visible and infrared transient absorption spectroscopies. The negative solvatochromic behavior of pyridinium phenolate indicates that the permanent electric dipole moment experiences a decrease upon S1 â S0 excitation, implying that the ground state possesses more zwitterionic character than the excited state. In contrast, pyrylium phenolate exhibits a weakly positive solvatochromic behavior corresponding to a small increase in the dipole moment upon excitation, implying more zwitterionic character in the excited than the ground state. Both compounds are therefore situated at different sides of the cyanine-limit structure, which has equally polar ground and excited states. Despite these differences, both molecules exhibit qualitatively similar excited-state properties. They are characterized by a very short fluorescence lifetime, increasing from about 1 to 20 ps, when varying solvent viscosity from 0.4 to 11 cP. There are, however, characteristic differences between the two compounds: The excited-state lifetimes of the pyrylium dye are shorter and also depend somewhat on polarity. The ensemble of spectroscopic data can be explained with a model where the emitting FranckâCondon excited state relaxes upon twisting around the single bond between the aryl units to a point where the excited- and ground-state surfaces are very close or intersect. After internal conversion to the ground state, the distorted molecule relaxes back to its equilibrium planar configuration, again largely dependent upon solvent viscosity. However, in this case, the kinetics for the pyrylium dye are slower than for the pyridinium dye and the polar solvent-induced acceleration is significantly stronger than in the excited state. This difference of kinetic behavior between the two compounds is a direct consequence of the change of the electronic structure from anormal to an overcritical merocyanine evidenced by steady-state spectroscopy. | ||||||||
 |
|
|||||||
The excited-state dynamics of the cationic dye malachite green (MG) and of the dianionic dye eosin B at the dodecane/water interface has been investigated using femtosecond time-resolved surface second harmonic generation (TR-SSHG). By using different probe wavelengths, the contributions of monomeric and aggregated MG to the signal could be spectroscopically distinguished. The effect of the addition of a small amount of surfactants was found to strongly depend on the relative charges of surfactant and dye. For surfactant/dye pairs with opposite charges, the TR-SSHG signal is dominated by the contribution from aggregates, whereas for pairs with the same charges, the signal intensity becomes vanishingly small. These effects are explained in terms of electrostatic interactions between surfactants and dyes that favor either attraction of the dye toward the interface or its repulsion toward the bulk. As a very similar behavior is observed with MG upon addition of NaSCN, we conclude that, in this case, this effect reflects the affinity of SCN¯ for the interface. On the other hand, the guanidinium cation was found to have a different effect than that of a positively charged surfactant on the SSHG signal of MG, indicating this cation does not accumulate in the interfacial region. | ||||||||
|
||||||||
Acylgermanes have been shown to act as efficient photoinitiators. In this investigation we show how dibenzoyldiethylgermane 1 reacts upon photoexcitation. Our real-time investigation utilizes femto- and nanosecond transient absorption, time-resolved EPR (50 ns), photo-chemically induced dynamic nuclear polarization, DFT calculations, and GC-MS analysis. The benzoyldiethylgermyl radical G⢠is formed via the triplet state of parent 1. On the nanosecond time scale this radical can recombine or undergo hydrogen-transfer reactions. Radical G⢠reacts with butyl acrylate at a rate of 1.2 ± 0.1 à 108 and 3.2 ± 0.2 à 108 Mâ1 sâ1, in toluene and acetonitrile, respectively. This is Ë1 order of magnitude faster than related phosphorus-based radicals. The initial germyl and benzoyl radicals undergo follow-up reactions leading to oligomers comprising GeâO bonds. LC-NMR analysis of photocured mixtures containing 1 and the sterically hindered acrylate 3,3-dimethyl-2-methylenebutanoate reveals that the products formed in the course of a polymerization are consistent with the intermediates established at short time scales. | ||||||||
|
 |
|||||||
We describe the experimental investigation of time-resolved magnetic field effects in exciplex-forming organic donorâacceptor systems. In these systems, the photoexcited acceptor state is predominantly deactivated by bimolecular electron transfer reactions (yielding radical ion pairs) or by direct exciplex formation. The delayed fluorescence emitted by the exciplex is magnetosensitive if the reaction pathway involves loose radical ion pair states. This magnetic field effect results from the coherent interconversion between the electronic singlet and triplet radical ion pair states as described by the radical pair mechanism. By monitoring the changes in the exciplex luminescence intensity when applying external magnetic fields, details of the reaction mechanism can be elucidated. In this work we present results obtained with the fluorophore-quencher pair 9,10-dimethylanthracene/N,N-dimethylaniline (DMA) in solvents of systematically varied permittivity. A simple theoretical model is introduced that allows discriminating the initial state of quenching, viz., the loose ion pair and the exciplex, based on the time-resolved magnetic field effect. The approach is validated by applying it to the isotopologous fluorophore-quencher pairs pyrene/DMA and pyrene-d10/DMA. We detect that both the exciplex and the radical ion pair are formed during the initial quenching stage. Upon increasing the solvent polarity, the relative importance of the distant electron transfer quenching increases. However, even in comparably polar media, the exciplex pathway remains remarkably significant. We discuss our results in relation to recent findings on the involvement of exciplexes in photoinduced electron transfer reactions. | ||||||||
|
||||||||
Modern spectroscopic techniques such as time-resolved second-harmonic-generation spectroscopy allow molecules to be examined selectively directly at phase interfaces. Two-phase systems formed by glycerol/water and alkane layers have previously been studied by time-resolved second-harmonic-generation spectroscopic measurements. In this molecular dynamics study, a triphenylmethane dye was inserted at the glycerol/waterâalkane interface and was used as a probe for local properties such as viscosity. We now show how extensive simulations over a wide range of concentrations can be used to obtain a detailed view of the molecular structure at the glycerol/waterâalkane interface. Glycerol is accumulated in a double layer adjacent to the alkane interface, which results in increased viscosity of the glycerol/water phase in the direct vicinity of the interface. We also show that conformational ensembles created by classical molecular-dynamics simulations can serve as input for QM/MM calculations, yielding further information such as transition dipoles, which can be compared with spectroscopic measurements. | ||||||||
 |
|
|||||||
Ultrafast transient absorption spectroscopy serves to identify the 3dd state as intermediate quencher state of the 3MLCT luminescence in the non-luminescent ruthenium complexes [Ru(m-bpy)3]2+ (m-bpy = 6-methyl-2,2â²-bipyridine) and [Ru(tm-bpy)3]2+ (tm-bpy = 4,4â²,6,6â²-tetramethyl-2â²,2â²-bipyridine). For [Ru(m-bpy)3]2+, the population of the 3dd state from the 3MLCT state occurs within 1.6 ps, while the return to the ground state takes 450 ps. For [Ru(tm-bpy)3]2+, the corresponding values are 0.16 and 7.5 ps, respectively. According to DFT calculations, methyl groups added in the 6 and 6â² positions of bipyridine stabilize the 3dd state by â¼4000 cmâ1 each, compared to [Ru(bpy)3]2+. | ||||||||
|
 |
|||||||
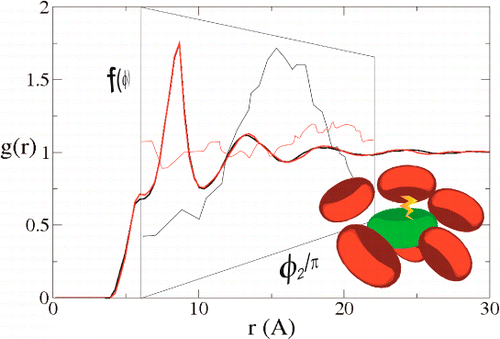
A major problem in the extraction of the reaction probability in bimolecular processes is the disentanglement from the influence of molecular diffusion. One of the strategies to overcome it makes use of reactive solvents in which the reactants do not need to diffuse to encounter each other. However, most of our quantitative understanding of chemical reactions in solution between free partners is based on the assumption that they can be approximated by spheres because rotation averages their mutual orientations. This condition may not be fulfilled when the reaction takes place on time scales faster than that of molecular reorientation. In this work, the fluorescence quenching of two very similar polyaromatic hydrocarbons with different electric dipole moments is measured. The concentration of a liquid electron-donating quencher is varied from very dilute solutions to pure quencher solutions. In both cases, the thermodynamics of the reactions are very similar and, according to the Marcus expression, the kinetics are expected to proceed at similar rates. However, one of them is 10 times faster in the pure quencher solution. This difference starts at relatively low quencher concentrations. An explanation based on the fluorophoreâsolvent dipoleâdipole interaction and the consequent orientational solvent structure is provided. The orientational correlation between fluorophore and quencher is calculated by means of computer simulations. Important differences depending on the fluorophore dipole moment are found. The kinetics can be explained quantitatively with a reactionâdiffusion model that incorporates the effects of the presence of the dipole moment and the rotational diffusion, only in the highest quencher concentration case, but not in dilute solutions, most likely due to fundamental limitations of the kinetic theory. | ||||||||
 |
|
|||||||
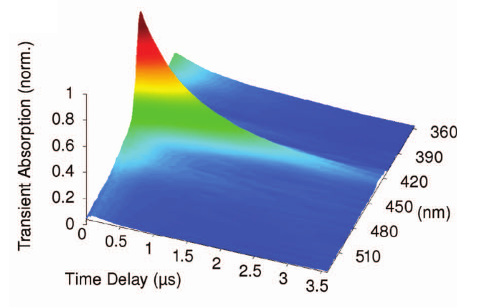
A combination of sub-nanosecond photoexcitation and femtosecond supercontinuum probing is used to extend femtosecond transient absorption spectroscopy into the nanosecond to microsecond time domain. Employing a passively Q-switched frequency tripled Nd:YAG laser and determining the jitter of the time delay between excitation and probe pulses with a high resolution time delay counter on a single-shot basis leads to a time resolution of 350 ps in picosecond excitation mode. The time overlap of almost an order of magnitude between fs and sub-ns excitation mode permits to extend ultrafast transient absorption (TA) experiments seamlessly into time ranges traditionally covered by laser flash photolysis. The broadband detection scheme eases the identification of intermediate reaction products which may remain undetected in single-wavelength detection flash photolysis arrangements. Single-shot referencing of the supercontinuum probe with two identical spectrometer/CCD arrangements yields an excellent signal-to-noise ratio for the so far investigated chromophores in short to moderate accumulation times. | ||||||||
|
 |
|||||||
Unambiguous evidence for the formation of excited ions upon ultrafast bimolecular photoinduced charge separation is found using a combination of femtosecond time-resolved fluorescence up-conversion, infrared and visible transient absorption spectroscopy. The reaction pathways are tracked by monitoring the vibrational energy redistribution in the product after charge separation and subsequent charge recombination. For moderately exergonic reactions, both donor and acceptor are found to be vibrationally hot, pointing to an even redistribution of the energy dissipated upon charge separation and recombination in both reaction partners. For highly exergonic reactions, the donor is very hot, whereas the acceptor is mostly cold. The asymmetric energy redistribution is due to the formation of the donor cation in an electronic excited state upon charge separation, confirming one of the hypotheses for the absence of the Marcus inverted region in photoinduced bimolecular charge separation processes | ||||||||
|
|
||||||||
Ultrafast photochemical processes can occur in parallel with the relaxation of the optically populated excited state toward equilibrium. The latter involves both intra- and intermolecular modes, namely vibrational and solvent coordinates, and takes place on timescales ranging from a few tens of femtoseconds to up to hundreds of picoseconds, depending on the system. As a consequence, the reaction dynamics can substantially differ from those usually measured with slower photoinduced processes occurring from equil-ibrated excited states. For example, the decay of the excited-state population may become strongly nonexponential and depend on the excitation wavelength, contrary to the Kasha and Vavilov rules. In this article, we first give a brief account of our current understanding of vibrational and solvent relaxation processes. We then present an overview of important classes of ultrafast photochemical reactions, namely electron and proton transfer as well as isomerization, and illustrate with several examples how nonequilibrium effects can affect their dynamics. | ||||||||
 |
|
|||||||
The interaction of a series of chiral cationic [4]helicene derivatives, which differ by their substituents, with double-stranded DNA has been investigated by using a combination of spectroscopic techniques, including time-resolved fluorescence, fluorescence anisotropy, and linear dichroism. Addition of DNA to helicene solutions results to a hypochromic shift of the visible absorption bands, an increase of fluorescence quantum yield and lifetime, a slowing down of fluorescence anisotropy decay, and a linear dichroism in flow-oriented DNA, which unambiguously points to the binding of these dyes to DNA. Both helicene monomers and dimeric aggregates, which form at higher concentration, bind to DNA, the former most probably upon intercalation and the latter upon groove binding. The binding constant depends substantially on the dye substituents and is, in all cases, larger with the M than the P enantiomer, by factors ranging from 1.2 to 2.3, depending on the dye. | ||||||||
|
||||||||
The time resolution of photon detection systems is important for a wide range of applications in physics and chemistry. It impacts the quality of time-resolved spectroscopy of ultrafast processes and has a direct influence on the best achievable time resolution of time-of-flight detectors in high-energy and medical physics. For the characterization of photon detectors, it is important to measure their exact timing properties in dependence of the photon flux and the operational parameters of the photodetector and its accompanying electronics. We report on the timing of silicon photomultipliers (SiPM) as a function of their bias voltage, electronics threshold settings and the number of impinging photons. We used ultrashort laser pulses at 400 nm wavelength with pulse duration below 200 fs. We focus our studies on different types of SiPMs (Hamamatsu MPPC S10931-025P, S10931-050P and S10931-100P) with different SPAD sizes (25μm, 50μm and 100μm) coupled to the ultrafast discriminator amplifier NINO. For the SiPMs, an optimum in the time resolution regarding bias and threshold settings can be reached. For the 50μm type, we achieve a single photon time resolution of 80 ps sigma, and for saturating photon fluxes better than 10 ps sigma. | ||||||||
|
||||||||
The photophysics and photochemistry of kynurenic acid (KNA) and kynurenine yellow (KNY) in neutral aqueous solutions were investigated using time-resolved optical spectroscopy. Both molecules have similar quinoline-like structures, the only difference being the absence of conjugation in the nitrogen containing cycle in KNY. The main channel of S1 excited state decay in the case of partially-unconjugated KNY is the solvent assisted S1 â S0 radiationless transition via intermolecular hydrogen bonds (ΦIC = 0.96), whereas, in the case of fully-conjugated KNA, it is intersystem crossing to the triplet state (ΦT = 0.82). The major intermediate products of the singlet excited KNY deactivation are the triplet state (ΦT = 0.022) and, most probably, the enol form (Φenol = 0.012), which decay with the formation of 2,3-dihydro-4-hydroxyquinoline and 4-hydroxyquinoline, respectively. The results obtained show that KNA and KNY, which are products of the decomposition of the UV filter kynurenine, are significantly more photoactive and less photostable than the parent molecule. | ||||||||
|
||||||||
The development of practical two-photon absorption photoinitiators (TPA PIs) has been slow due to their complicated syntheses often reliant on expensive catalysts. These shortcomings have been a critical obstruction for further advances in the promising field of two-photon-induced photopolymerization (TPIP) technology. This paper describes a series of linear and cyclic benzylidene ketone-based two-photon initiators containing double bonds and dialkylamino groups synthesized in one step via classical aldol condensation reactions. Systematic investigations of structureâactivity relationships were conducted via quantum-chemical calculations and experimental tests. These results showed that the size of the central ring significantly affected the excited state energetics and emission quantum yields as well as the two-photon initiation efficiency. In the TPIP tests the 4-methylcyclohexanone-based initiator displayed much broader ideal processing windows than its counterparts with a central five-membered ring and previously described highly active TPA PIs. Surprisingly, a writing speed as high as 80 mm/s was obtained for the microfabrication of complex 3D structures employing acrylate-based formulations. These highly active TPA PIs also exhibit excellent thermal stability and remain inert to one-photon excitation. Straightforward synthesis combined with high TPA initiation efficiency makes these novel initiators promising candidates for commercialization. | ||||||||
|
 |
|||||||
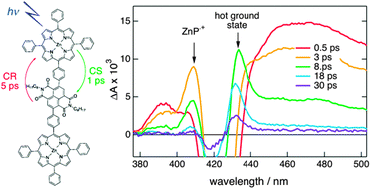
The excited-state dynamics of two energy donorâbridgeâacceptor (DâBâA) systems consisting of a zinc tetraphenylporphyrin (ZnP) and a free base tetraphenylporphyrin (FbP) bridged by oligo-p-phenyleneethynylene units with different substituents has been investigated using ultrafast spectroscopy. These systems differ by the location of the lowest singlet excited state of the bridge, just above or below the S2Â porphyrin states. In the first case, Soret band excitation of the porphyrins is followed by internal conversion to the local S1Â state of both molecules and by a S1Â energy transfer from the ZnP to the FbP end on the 10 ns time scale, as expected for a center-to-center distance of about 4.7 nm. On the other hand, if the bridge is excited, the energy is efficiently transferred within 1 ps to both porphyrin ends. Selective bridge excitation is not possible with the second system, because of the overlap of the absorption bands. However, the time-resolved spectroscopic data suggest a reversible conversion between the D*(S2)âBâA and DâB*(S1)âA states as well as a transition from the DâB*(S1)âA to the DâBâA* states on the picosecond time scale. This implies that the local S2energy of the ZnP end can be transported stepwise to the FbP end, i.e., over about 4.7 nm, within 1 ps with an efficiency of more than 0.2. | ||||||||
 |
|
|||||||
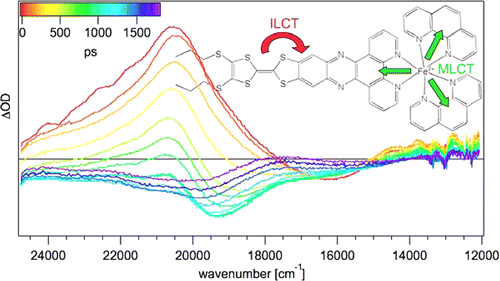
The synthesis and photophysical properties of the complex [Fe(phen)2(TTF-dppz)]2+Â (TTF-dppz = 4â²,5â²-bis-(propylthio)tetrathiafulvenyl[i]dipyrido[3,2-a:2â²,3â²-c]phenazine, phen = 1,10-phenanthroline) are described. In this complex, excitation into the metalâligand charge transfer bands results in the population of a high-spin state of iron(II), with a decay lifetime of approximately 1.5 ns, in dichloromethane, at room temperature. An intraligand charge transfer state can also be obtained and has a lifetime of 38 ps. A mechanism for the different states reached is proposed based on transient absorption spectroscopy. | ||||||||
|
 |
|||||||
The excited-state dynamics of two triads consisting of a naphthalenediimide (cNDI) substituted at the core by two zinc (ZnP) or free-base tetraphenylporphyrins (FbP) was investigated by ultrafast fluorescence and transient absorption spectroscopy. The electronic absorption spectra of the triads are almost the composites of those of the constituents, pointing to a weak electronic coupling and to a localization of the excitation energy on one of the porphyrins. In cyclohexane, the excited-state dynamics of the triads are essentially the same as those of the individual porphyrins, with the exception of the Soret emission of the ZnP triad, whose lifetime exhibits a more than 10 fold shortening compared to ZnP. A similarly ultrafast fluorescence decay was measured in tetrahydrofuran and benzonitrile. In these two solvents, charge separation from the excited porphyrin to the cNDI was found to take place with ~1 ps and ~25 ps time constants in the ZnP and FbP triads, respectively. The build up of the charge-separated state population in the ZnP triad is independent on the excitation wavelength, indicating that charge separation takes place from the lowest singlet excited state. Charge recombination occurs with a time constant around 8 ps in both triads, i.e. is slower than charge separation in the ZnP triad but faster in the FbP triad. These differences are rationalized in terms of the driving forces for charge separation and recombination. | ||||||||
|
||||||||
The synthesis, X-ray structures and photophysical properties of several new Ln(III) complexes with pyrazine-2,6-dicarboxylic acid (H2PYZ) that demonstrate excellent stability and solubility in non-aqueous solution are reported, and compared to structurally analogous complexes with pyridine-2,6-dicarboxylic acid (H2DPA). The Eu(III) and Yb(III) complexes demonstrate efficient metal centered luminescence in the visible and Near Infra-Red (NIR) regions respectively. Low temperature (77 K) phosphorescence measurements using the corresponding Gd(III) complex allowed the photophysical properties of the sensitizer to be rationalized, together with corresponding TD-DFT studies for a model complex. Lastly, we have evaluated the sensitization efficiencies for these complexes, and have undertaken femtosecond transient absorption (TA) measurements in order to evaluate the relative importance of the intersystem crossing and energy transfer processes involved with sensitized Ln(III) emission via the antennae effect. | ||||||||

){kind=link}
Download this list in a RIS file or a BIB file or a PDF file
Contact:
Eric Vauthey
Physical Chemistry Department - Sciences II - University of Geneva
30, Quai Ernest Ansermet - CH-1211 Geneva 4 (Switzerland)
© All rights reserved by Eric Vauthey and the University of Geneva
Design and code by Guillaume Duvanel