

« Back to all publications
Download this list in a RIS file or a BIB file or a PDF file
|
||||||||
The objective with synthetic multifunctional nanoarchitecture is to create large suprastructures with interesting functions. For this purpose, lipid bilayer membranes or conducting surfaces have been used as platforms and rigid-rod molecules as shape-persistent scaffolds. Examples for functions obtained by this approach include pores that can act as multicomponent sensors in complex matrices or rigid-rod π-stack architecture for artificial photosynthesis and photovoltaics. | ||||||||



|
||||||||
Polarization-sensitive ultrafast infrared measurements on photoinduced electron transfer in donor-acceptor pairs in polar acetonitrile show distinct contributions from loose and tight ion pairs. Highly anisotropic signals from tight ion pairs reveal the importance of mutual orientation of the reactants (see picture) and thus the need to refine theoretical models based on spherical species that solely involve reaction distances. | ||||||||
|
 |
|||||||
The excited-state dynamics of covalently linked electron donor−acceptor systems consisting of N,N-dimethylaniline (DMA) as electron donor and either perylene (Pe) or cyanoperylene (CNPe) as acceptor has been investigated in a large variety of solvents, including a room-temperature ionic liquid, by using femtosecond time-resolved fluorescence and absorption spectroscopy. The negligibly small solvent dependence of the absorption spectrum of both compounds and the strong solvatochromism of the fluorescence are interpreted by a model where optical excitation results in the population of a locally excited state (LES) and emission takes place from a charge-separated state (CSS). This interpretation is supported by the fluorescence up-conversion and the transient absorption measurements that reveal substantial spectral dynamics in polar solvents only, occurring on time scales going from a few hundreds of femtoseconds in acetonitrile to several tens of picoseconds in the ionic liquid. The early transient absorption spectra are similar to those found in nonpolar solvents and are ascribed to the LES absorption. The late spectra due to CSS absorption show bands that are red-shifted relative to those of the radical anion of the acceptor moiety by an amount that depends on solvent polarity, pointing to partial charge separation. Global analysis of the time-resolved data indicates that the charge separation dynamics in PeDMA is essentially solvent controlled, whereas that in CNPeDMA is faster than diffusive solvation, this difference being accounted for by a larger driving force for charge separation in the latter. On the other hand, the CSS lifetime of PeDMA is of the order of a few nanoseconds independently of the solvent, whereas that of CNPeDMA decreases with increasing solvent polarity from a few nanoseconds to a few hundreds of picoseconds. Comparison of these results with previously published data on the fluorescence quenching of Pe and CNPe in pure DMA shows that the charge separation and the ensuing charge recombination occur on similar time scales independently of whether these processes are intra- or intermolecular. | ||||||||
 |
|
|||||||
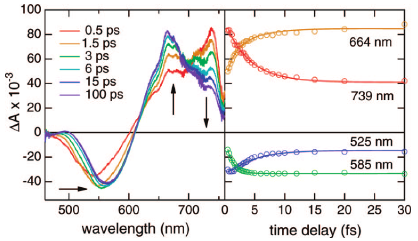
The photophysical properties of multichromophoric systems consisting of eight red or blue naphthalene diimides (NDIs) covalently attached to a p-octiphenyl scaffold, as well as a blue bichromophoric system with a biphenyl scaffold, have been investigated in detail using femtosecond time-resolved spectroscopy. The blue octachromophoric systems have been recently shown to self-assemble as supramolecular tetramers in lipid bilayer membranes and to enable generation of a transmembrane proton gradient upon photoexcitation (Bhosale, S.; Sisson, A. L.; Talukdar, P.; Fürstenberg, A.; Banerji, N.; Vauthey, E.; Bollot, G.; Mareda, J.; Röger, C.; Würthner, F.; Sakai, N.; Matile, S. Science2006, 313, 84). A strong reduction of the fluorescence quantum yield was observed when going from the single NDI units to the multichromophoric systems in methanol, the effect being even stronger in a vesicular lipid membrane. Fluorescence up-conversion measurements reveal ultrafast self-quenching in the multichromophoric systems, whereas the formation of the NDI radical anion, evidenced by transient absorption measurements, points to the occurrence of photoinduced charge separation. The location of the positive charge could not be established unambiguously from the transient absorption measurements, but energetic considerations indicate that charge separation should occur between two NDI units in the blue systems, whereas both an NDI unit and the p-octiphenyl scaffold could act as electron donor in the red system. The lifetime of the charge-separated state was found to increase from 22 to 45 ps by going from the bi- to the octachromophoric blue systems in methanol, while a 400 ps decay component was observed in the lipid membrane. This lifetime lengthening is explained in terms of charge migration that is most efficient when the octachromophoric systems are assembled as supramolecular tetramers in the lipid membrane. Furthermore, the average charge-separated state lifetime of the red system in methanol is even larger and amounts to 750 ps. This effect cannot be simply explained in terms of Marcus inverted regime as the driving force for charge recombination in the red system is only slightly larger than in the blue one. A better spatial separation of the charges in the red system stemming from the localization of the hole on the p-octiphenyl scaffold could additionally contribute to the slowing down of charge recombination. | ||||||||
|
 |
|||||||
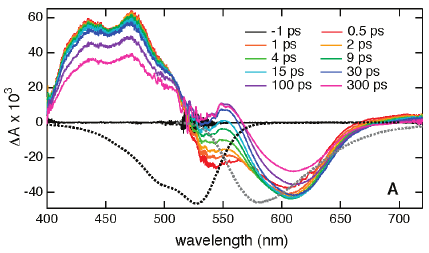
The excited-state dynamics of the methylperylene/tetracyanoethylene (MPe/TCNE) donor−acceptor complex has been investigated in various solvents using femtosecond transient absorption spectroscopy. The transient spectra reveal the formation of two types of ion pairs: The first (IP1), constituting the major fraction of the total ion-pair population, is characterized by a broad and red-shifted absorption spectrum compared to that of the free MPe cation and by a subpicosecond lifetime, whereas the second (IP2) has a spectrum closer to that of MPe cation and a lifetime of a few picoseconds. A substantial polarization anisotropy was observed with IP1 but not with IP2, indicating a relatively well-defined structure for the former. The reaction scheme that best accounts for the observed dynamics and its solvent dependence involves the simultaneous excitation of complexes that differ by their electronic coupling. The more coupled complexes have a high absorption coefficient and thus yield IP1, which undergoes ultrafast charge recombination, whereas the less coupled complexes have a lower probability to be excited and lead to the longer-lived IP2. | ||||||||
 |
|
|||||||
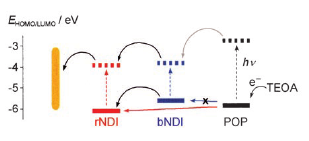
Supramolecular 3D organization on gold with interdigitating intra- and interlayer recognition motifs (see picure, black p-oligophenyl rods; red, blue naphthalenediimide (NDI) stacks) is designed to access supramolecular cascade n/p-heterojunctions or the adaptable directionality needed to control fill factors in current-voltage curves. | ||||||||
|
 |
|||||||
The photophysics and excited-state dynamics of nitroperylene (NPe) in solvents of various polarities and viscosities, including a room-temperature ionic liquid, have been investigated by femtosecond-resolved transient absorption spectroscopy. The excited-state absorption spectrum was found to depend substantially on solvent polarity. In the most polar solvents, it is very similar to that of the NPe radical cation generated upon bimolecular quenching by an electron acceptor, denoting a substantial charge-transfer character of the S1 state. Contrary to smaller nitroaromatic compounds, NPe in the S1 state does not undergo ultrafast intersystem crossing (ISC) but decays mainly by internal conversion (IC). In nonprotic solvents, IC involves low-frequency modes with large amplitude motion associated with the nitro group and depends on both the solvent viscosity and polarity. It takes place on a 100 ps time scale in acetonitrile, while in cyclohexane, it is slow enough for ISC to become competitive. Moreover, both the fluorescence quantum yield and the excited-state dynamics were found to differ, depending on which side of the S0−S1 absorption band excitation was performed. This dependence is explained by the inhomogeneous nature of the absorption spectrum arising from a distribution of twist angles of the nitro group relative to the aromatic plane. On the other hand, such excitation wavelength effects were not observed in protic solvents, where the excited-state lifetime was found to be substantially shorter than that in nonprotic solvents. This behavior is rationalized in terms of a H-bonding interaction, which limits the torsional disorder of NPe and favors ultrafast nonradiative deactivation of the excited state. Transient absorption measurements performed for comparative purpose with nitropyrene in acetonitrile confirm the occurrence of ultrafast ISC in smaller nitroaromatic compounds. | ||||||||
|
||||||||
The charge recombination dynamics of excited donor−acceptor complexes consisting of hexamethylbenzene (HMB), pentamethylbenzene (PMB), and isodurene (IDU) as electron donors and tetracyanoethylene (TCNE) as electron acceptor in various polar solvents has been investigated within the framework of the stochastic approach. The model accounts for the reorganization of intramolecular high-frequency vibrational modes as well as for the solvent reorganization. All electron-transfer energetic parameters have been determined from the resonance Raman data and from the analysis of the stationary charge transfer absorption band, while the electronic coupling has been obtained from the fit to the charge recombination dynamics in one solvent. It appears that nearly 100% of the initially excited donor−acceptor complexes recombine in a nonthermal (hot) stage when the nonequilibrium wave packet passes through a number of term crossings corresponding to transitions toward vibrational excited states of the electronic ground state. Once all parameters of the model have been obtained, the influence of the dynamic solvent properties (solvent effect) and of the carrier frequency of the excitation pulse (spectral effect) on the charge recombination dynamics have been explored. The main conclusions are (i) the model provides a globally satisfactory description for the IDU/TCNE complex although it noticeably overestimates the spectral effect, (ii) the solvent effect is quantitatively well described for the PMB/TCNE and HMB/TCNE complexes but the model fails to reproduce their spectral effects, and (iii) the positive spectral effect observed with the HMB/TCNE complex cannot be described within the framework of two-level models and the charge redistribution in the excited complexes should most probably be taken into account. | ||||||||
Download this list in a RIS file or a BIB file or a PDF file
Contact:
Eric Vauthey
Physical Chemistry Department - Sciences II - University of Geneva
30, Quai Ernest Ansermet - CH-1211 Geneva 4 (Switzerland)
© All rights reserved by Eric Vauthey and the University of Geneva
Design and code by Guillaume Duvanel