

« Back to all publications
Download this list in a RIS file or a BIB file or a PDF file
|
 |
|||||||
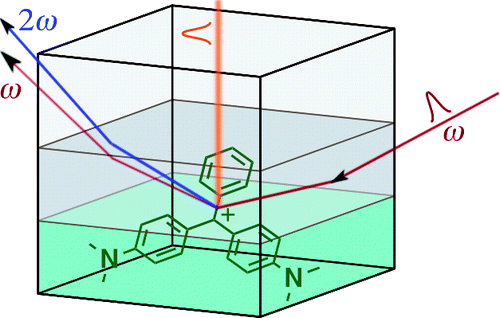
The excited-state relaxation of malachite green and brilliant green in solvents of various viscosity has been investigated at liquid/liquid interfaces and in bulk solutions by surface second harmonic generation and transient absorption spectroscopy. Mixtures of water and glycerol in various proportions have been used as solvents of variable viscosity. Transient absorption measurements in bulk revealed that both dyes are suitable as a probe of local viscosity for water+glycerol mixtures and that two of three processes following the optical excitation exhibit the same power dependence on solvent viscosity. This observation leads to assignment of the processes to a twist and twist-back of the aromatic rings attached to the central carbon atom of the dye. Therefore, identification of the intermediate state observed in the radiationless deactivation pathway with the twisted form of the dye has been supported. The time profiles of the second harmonic signal recorded at water+glycerol/dodecane interfaces have been found to be monoexponential at low dye concentrations (below 10-5 M) and biexponential at higher concentrations, and therefore the origin of the slower component has been attributed to the relaxation of dye aggregates adsorbed at the interface. The decay times measured at interfaces increased with increasing amount of glycerol in the mixture, but the rise was slower than in bulk solution. Therefore, the viscosity at the interfacial region, higher than that of the bulk solution, is mainly determined by structural modification of the solvent resulting from interactions between the two liquids that constitute the interface and addition of glycerol affects viscosity, only to a lesser extent. We have also shown that if the viscosity of the upper layer is much higher (at least 1 order of magnitude) than that of water or short alkanes, a slow-down of the relaxation is observed. This contradicts earlier findings and means that large amplitude motion of all three rings is involved in the deactivation of the excited molecule, but the rotation of the phenyl ring, which is smaller than the alkyl-substituted aniline groups, becomes a bottleneck for the relaxation in very viscous environments. | ||||||||



 |
|
|||||||
Matching matters when building supramolecular n/p-heterojunction photosystems on solid supports that excel with efficient photocurrent generation, important critical thickness, smooth surfaces, and flawless responsiveness to functional probes for the existence of operational intra- and interlayer recognition motifs. | ||||||||
|
||||||||
In this study, we describe synthesis, characterization, and zipper assembly of yellow p-oligophenyl naphthalenediimide (POP-NDI) donor−acceptor hybrids. Moreover, we disclose, for the first time, results from the functional comparison of zipper and layer-by-layer (LBL) assembly as well as quartz crystal microbalance (QCM), atomic force microscopy (AFM), and molecular modeling data on zipper assembly. Compared to the previously reported blue and red NDIs, yellow NDIs are more π-acidic, easier to reduce, and harder to oxidize. The optoelectronic matching achieved in yellow POP-NDIs is reflected in quantitative and long-lived photoinduced charge separation, comparable to their red and much better than their blue counterparts. The direct comparison of zipper and LBL assemblies reveals that yellow zippers generate more photocurrent than blue zippers as well as LBL photosystems. Continuing linear growth found in QCM measurements demonstrates that photocurrent saturation at the critical assembly thickness occurs because more charges start to recombine before reaching the electrodes and not because of discontinued assembly. The found characteristics, such as significant critical thickness, strong photocurrents, large fill factors, and, according to AFM images, smooth surfaces, are important for optoelectronic performance and support the existence of highly ordered architectures. | ||||||||
|
 |
|||||||
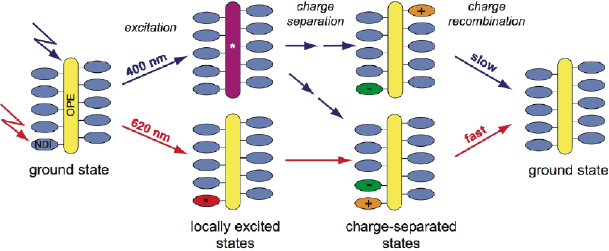
The photophysical properties of two hybrid multichromophoric systems consisting of an oligophenylethynyl (OPE) scaffold decorated by 10 red or blue naphthalene diimides (NDIs) have been investigated using femtosecond spectroscopy. Ultrafast charge separation was observed with both red and blue systems. However, the nature of the charge-separated state and its lifetime were found to differ substantially. For the red system, electron transfer occurs from the OPE scaffold to an NDI unit, independently of whether the OPE or an NDI is initially excited. However, charge separation upon OPE excitation is about 10 times faster, and takes place with a 100 fs time constant. The average lifetime of the ensuing charge-separated state amounts to about 650 ps. Charge separation in the blue system depends on which of the OPE scaffold or an NDI is excited. In the first case, an electron is transferred from the OPE to an NDI and the hole subsequently shifts to another NDI unit, whereas in the second case symmetry-breaking charge separation between two NDI units occurs. Although the charges are located on two NDIs in both cases, different recombination dynamics are observed. This is explained by the location of the ionic NDI moieties that depends on the charge separation pathway, hence on the excitation wavelength. The very different dynamics observed with red and blue systems can be accounted for by the oxidation potentials of the respective NDIs that are higher and lower than that of the OPE scaffold. Because of this, the relative energies of the two charge-separated states (hole on the OPE or an NDI) are inverted. | ||||||||
|
||||||||
The ultrafast excited-state dynamics of Malachite Green (MG) in bulk aqueous solutions and at air/water interfaces, in particular the effect of the presence of various sodium salts in the aqueous phase, has been investigated by transient absorption and surface second harmonic generation. In bulk solutions, a slowing down of the ground-state recovery that can be unambiguously ascribed to the formation of aggregates of various sizes is observed at high (>0.3 M) salt concentrations only, with the exception of NaSCN where an effect is already found at 0.05 M. At the interface, small amounts of salt result in two effects: 1) an increase of the stationary surface second harmonic signal and 2) a slowing down of the ground-state recovery of MG. These phenomena are explained by the formation of aggregates due to an increase of the interfacial MG concentration upon addition of salt. The dependencies of both effects on salt concentration are correlated and vary with the anion as SCN− > Br− > SO4− > Cl−. This order is almost the opposite as that in the Hofmeister series for the salting-out strength. | ||||||||
 |
|
|||||||
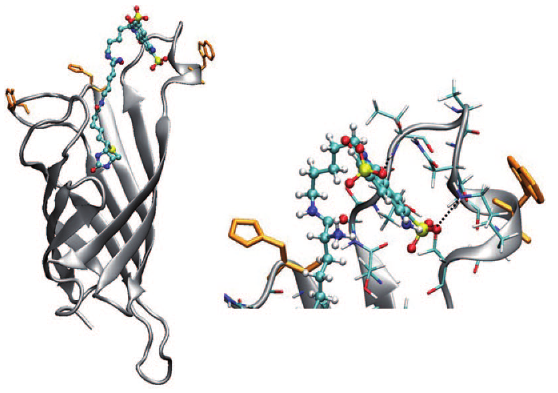
The excited-state dynamics of biotin–spacer–Lucifer-Yellow (LY)constructs bound to avidin (Avi) and streptavidin (Sav) was investigatedusing femtosecond spectroscopy. Two different locations in the proteins,identified by molecular dynamics simulations of Sav, namely the entrance of the binding pocket andthe protein surface, were probed by varying the length of thespacer. A reduction of the excited-state lifetime, stronger inSav than in Avi, was observed with the long spacer construct.Transient absorption measurements show that this effect originatesfrom an electron transfer quenching of LY, most probablyby a nearby tryptophan residue. The local environment of theLY chromophore could be probed by measuring the time-dependent polarisation anisotropy and Stokes shift of the fluorescence. Substantial differences in both dynamics were observed.The fluorescence anisotropy decays analysed by using thewobbling-in-a-cone model reveal a much more constrained environment of the chromophore with the short spacer. Moreover, the dynamic Stokes shift is multiphasic in all cases, with a~ 1 ps component that can be ascribed to diffusive motion ofbulk-like water molecules, and with slower components withtime constants varying not only with the spacer, but with theprotein as well. These slow components, which depend strongly on the local environment of the probe, are ascribed to themotion of the hydration layer coupled to the conformationaldynamics of the protein. | ||||||||
|
||||||||
The photophysics of a number salicylic acid derivatives (SADs) in aqueous solutions was investigated in a wide range of pH by time-correlated single photon counting (λex = 350 nm, τresp = 300 ps) and fluorescence up-conversion (λex = 266 nm, τresp = 300 fs) techniques. The acid-base equilibrium constants in the ground (pKa) and the excited states (pKa*), the fluorescence quantum yields as well as the lifetimes of anionic, neutral, and cationic forms of SADs were determined. Evidence of ultrafast excited-state intramolecular proton transfer (ESIPT) leading to the formation of the proton-transferred excited state of SADs was obtained from the fluorescence up-conversion measurement. The nature of the ESIPT process is discussed. | ||||||||
|
 |
|||||||
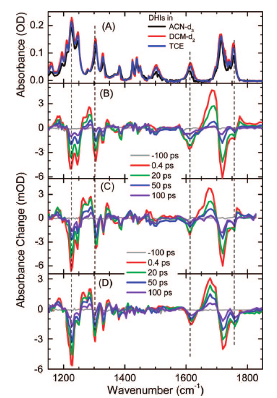
We present results of a femtosecond spectroscopy study of the ring-opening dynamics of the photochromiccompound trimethyl-1′H-spiro[fluorene-9,1′-pyrrolo[1,2-b]pyridazines]-2′,3′,6′-tricarboxylate (also known asdihydroindolizine and abbreviated as DHI) in solvents of different polarities. We follow the ring-openingdynamics of photoexcited DHI by probing the transient response in the visible region between 450 and 700nm, as well as in the fingerprint region between 1100 and 1800 cm-1. We conclude that photoexcited DHIconverts into the ring-opened betaine isomer while remaining in the electronic excited state. Subsequentelectronic excited-state decay on a time scale of 40-80 ps results in regeneration of ground-state DHI (0.75-0.9quantum yield) or betaine photoproduct, the exact value for DHI quantum yield recoveries and rates beingsolvent dependent. Figure Steady state of DHI in ACN-d3, DCM-d2, and TCE (A).Transient spectra of DHI at different pulse delays after 400 nm laserexcitation in ACN-d3 (B), in DCM-d2 (C), and in TCE (D). | ||||||||
 |
|
|||||||
The excited-state dynamics of kynurenine (KN) has been examined in various solvents by femtosecond-resolved optical spectroscopy. The lifetime of the S1 state of KN amounts to 30 ps in aqueous solutions, increases by more than 1 order of magnitude in alcohols, and exceeds 1 ns in aprotic solvents such as DMSO and DMF, internal conversion (IC) being shown to be the main deactivation channel. The IC rate constant is pH independent but increases with temperature with an activation energy of about 7 kJ/mol in all solvents studied. The dependence on the solvent proticity together with the observation of a substantial isotope effect indicates that hydrogen bonds are involved in the rapid nonradiative deactivation of KN in water. These results give new insight into the efficiency of KN as a UV filter and its role in cataractogenesis. | ||||||||
|
||||||||
Terms of donor-acceptor complex states participating in transitions with the excitation of the CT1 and CT2 bands (vertical arrows). The dotted arrow is the radiationless transition. Dotted lines are vibrational sublevels of the ground electronic state. Dashed arrows show the directions of system relaxation before and after nonthermal transitions. | ||||||||
|
 |
|||||||
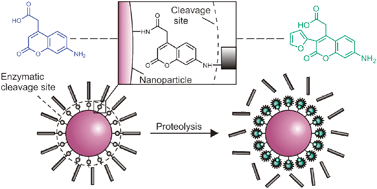
Protease responsive nanosensors were obtained by the attachment of unique green fluorescent bifunctional 3-arylcoumarin-derived fluorogenic substrates to poly(acrylamide-co-N-(3-aminopropyl)methacrylamide) nanoparticles, in which proteolysis results in substantial signal amplification. | ||||||||
 |
|
|||||||
The kinetics of the dual fluorescence of several derivatives of dimethylaminobenzonitrile (DMABN) has been compared using fs-fluorescence upconversion experiments. Variation of the size and twist angle of the donor (dialkylamino group) suggest a large amplitude solvent-viscosity controlled diffusional twisting motion towards larger twist angles as the rate limiting step. Large rate differences were observed for an ester group as acceptor. Temperature dependent studies indicate that these differences are not connected with different activation barriers but with changes in the Arrhenius preexponential factor. It is argued that conical intersections along the reaction path can bring about these entropy changes. | ||||||||
Download this list in a RIS file or a BIB file or a PDF file
Contact:
Eric Vauthey
Physical Chemistry Department - Sciences II - University of Geneva
30, Quai Ernest Ansermet - CH-1211 Geneva 4 (Switzerland)
© All rights reserved by Eric Vauthey and the University of Geneva
Design and code by Guillaume Duvanel