

|
||||||||
The self-assembly of core-substituted naphthalene diimides bearing triethylene glycol motifs leads to the formation of stable vesicles in DMSO and CHCl3/MeOH (6 : 4, v/v) solvents. The vesicles were evaluated by means of UV/vis and fluorescence spectroscopy, transmission electron microscopy, atomic force microscopy and dynamic light scattering. | ||||||||



|
||||||||
The green-fluorescent protein of the jellyfish operates with the most powerful phenolate donors in the push–pull fluorophore. To nevertheless achieve red fluorescence with the same architecture, sea anemone and corals apply oxidative imination, a process that accounts for the chemistry of vision as well. The objective of this study was to apply these lessons from nature to one of the most compact family of panchromatic fluorophores, i.e. core-substituted naphthalenediimides (cNDIs). We report straightforward synthetic access to hydroxylated cNDI and cPDI cores by palladium-catalyzed cleavage of allyloxy substituents. With hydroxylated cNDIs but not cPDIs in water-containing media, excited-state intramolecular proton transfer yields a second bathochromic emission. Deprotonation of hydroquinone, catechol and boronic ester cores provides access to an impressive panchromism up to the NIR frontier at 640 nm. With cNDIs, oxidative imination gives red shifts up to 638 nm, whereas the expanded cPDIs already absorb at 754 nm upon deprotonation of hydroquinone cores. The practical usefulness of hydroquinone cNDIs is exemplified by ratiometric sensing of the purity of DMF with the “naked eye” at a sensitivity far beyond the “naked nose”. We conclude that the panchromatic hypersensitivity toward the environment of the new cNDIs is ideal for pattern generation in differential sensing arrays. | ||||||||
|
 |
|||||||
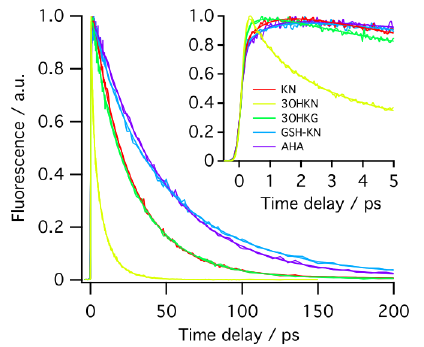
PURPOSE. To compare the photochemical properties of UV filter molecules present in the human lens (kynurenine, KN; 3-hy- droxykynurenine, 3OHKN; 3-hydroxykynurenine O-gluco- side, 3OHKG; 4-(2-aminophenyl)-4-oxobutanoic acid, AHA; and glutathionyl-kynurenine, GSH-KN) with the use of the following parameters: excited singlet lifetime, fluorescence quantum yield, triplet quantum yield, photodecomposition quantum yield. METHODS. The excited singlet lifetimes were measured with the use of fluorescence upconversion (time resolution, 210 fs) and pump-probe transient absorption (time resolution, 200 fs) methods. The fluorescence quantum yields were determined relative to an aqueous solution of quinine bisulfate. The triplet quantum yields were measured with the use of nanosecond laser flash photolysis. The photodecomposition quantum yields were determined by steady state photolysis followed by the high-performance liquid chromatography analysis. RESULTS. The secondary UV filters—AHA and GSH-KN are better photosensitizers than the primary ones -KN, 3OHKN and 3OHKG: the singlet state lifetimes of the secondary UV filters are longer, and the quantum yields of fluorescence and triplet state formation are higher. CONCLUSIONS. With aging, the ratio primary/secondary UV filters in the human lens decreases from approximately 10:1 to 2:1. The obtained results demonstrate that the quality of the secondary UV filters is inferior compared to the primary ones, which may result in a higher susceptibility of old lenses to UV light. That might be an important factor for the development of the age-related cataract. | ||||||||
|
||||||||
Facile access to complex systems is crucial to generate the functional materials of the future. Herein, we report self-organizing surface-initiated polymerization (SOSIP) as a user-friendly method to create ordered as well as oriented functional systems on transparent oxide surfaces. In SOSIP, self-organization of monomers and ring-opening disulfide exchange polymerization are combined to ensure the controlled growth of the polymer from the surface. This approach provides rapid access to thick films with smooth, reactivatable surfaces and long-range order with few defects and high precision, including panchromatic photosystems with oriented four-component redox gradients. The activity of SOSIP architectures is clearly better than that of disordered controls. | ||||||||
 |
|
|||||||
We present the femtosecond spectroscopic investigation of a covalently linked dyad, PCB-P3HT, formed by a segment of the conjugated polymer P3HT (regioregular poly(3-hexylthiophene)) that is end capped with the fullerene derivative PCB ([6,6]-phenyl-C61-butyric acid ester), adapted from PCBM. The fluorescence of the P3HT segment in tetrahydrofuran (THF) solution is reduced by 64% in the dyad compared to a control compound without attached fullerene (P3HT-OH). Fluorescence upconversion measurements reveal that the partial fluorescence quenching of PCB-P3HT in THF is multiphasic and occurs on an average time scale of 100 ps, in parallel to excited-state relaxation processes. Judging from ultrafast transient absorption experiments, the origin of the quenching is excitation energy transfer from the P3HT donor to the PCB acceptor. Due to the much higher solubility of P3HT compared to PCB in THF, the PCB-P3HT dyad molecules self-assemble into micelles. When pure C60 is added to the solution, it is incorporated into the fullerene-rich center of the micelles. This dramatically increases the solubility of C60 but does not lead to significant additional quenching of the P3HT fluorescence by the C60 contained in the micelles. In PCB-P3HT thin films drop-cast from THF, the micelle structure is conserved. In contrast to solution, quantitative and ultrafast (<150 fs) charge separation occurs in the solid-state films and leads to the formation of long-lived mobile charge carriers with characteristic transient absorption signatures similar to those that have been observed in P3HT:PCBM bulk heterojunction blends. While π -stacking interactions between neighboring P3HT chains are weak in the micelles, they are strong in thin films drop-cast from ortho-dichlorobenzene. Here, PCB-P3HT self-assembles into a network of long fibers, clearly seen in atomic force microscopy images. Ultrafast charge separation occurs also for the fibrous morphology, but the transient absorption experiments show fast loss of part of the charge carriers due to intensity-induced recombination and annihilation processes and monomolecular interfacial trap-mediated or geminate recombination. The yield of the long-lived charge carriers in the highly organized fibers is however comparable to that obtained with annealed P3HT:PCBM blends. PCB-P3HT can therefore be considered as an active material in organic photovoltaic devices. | ||||||||
|
 |
|||||||
Even flow: Photoinduced symmetry-breaking charge separation takes place in a few picoseconds in a 1,3-bis(perylene)propane dyad in polar solvents. Polarized transient absorption measurements show that the direction of the charge flow is random and entirely governed by the fluctuations of the solvent orientation around the dyad. | ||||||||
 |
|
|||||||
Several novel aromatic ketone-based two-photon initiators containing triple bonds and dialkylamino groups were synthesized and the structure-activity relationships were evaluated. Branched alkyl chains were used at the terminal donor groups to improve the solubility in the multifunctional monomers. Because of the long conjugation length and good coplanarity, the evaluated initiators showed large two-photon cross section values, while their fluorescence lifetimes and quantum yields strongly depend on the solvent polarity. All novel initiators exhibited high activity in terms of two-photon-induced microfabrication. This is especially true for fluorenone-based derivatives, which displayed much broader processing windows than well-known highly active initiators from the literature and commercially available initiators. While the new photoinitiators gave high reactivity in two-photon-induced photopolymerization at concentration as low as 0.1% wt, these compounds are surprisingly stable under one photon condition and nearly no photo initiation activity was found in classical photo DSC experiment. | ||||||||
|
|
||||||||
The p53 tumour suppressor gene, the most frequently mutated gene in human cancer, encodes a transcription factor that contains sequence-specific DNA binding and homo-tetramerization domains. Interestingly, the affinities of p53 for specific and non-specific DNA sites differ by only one order of magnitude, making it hard to understand how this protein recognizes its specific DNA targets in vivo. We describe here the structure of a p53 polypeptide containing both the DNA binding and oligomerization domains in complex with DNA. The structure reveals that sequence- specific DNA binding proceeds via an induced fit mechan- ism that involves a conformational switch in loop L1 of the p53 DNA binding domain. Analysis of loop L1 mutants demonstrated that the conformational switch allows DNA binding off-rates to be regulated independently of affinities. These results may explain the universal prevalence of conformational switching in sequence-specific DNA binding proteins and suggest that proteins like p53 rely more on differences in binding off-rates, than on differences in affinities, to recognize their specific DNA sites. | ||||||||
|
||||||||
The activities of our research group in the field of photoinduced electron transfer reactions are discussed and illustrated by several examples | ||||||||
|
 |
|||||||
The femtosecond-resolved evolution of the emission spectrum of the important conjugated polymer poly(3-hexylthiophene) (P3HT) is presented. Detailed fluorescence up-conversion spectroscopy was performed on P3HT solid-state films and on P3HT in chlorobenzene solution. Two excitation wavelengths and several emission wavelengths, covering the entire fluorescence spectrum, were used. The data were complemented by polarization-sensitive measurements. Our global analysis allowed a reconstruction of the time-resolved emission spectra with 200 fs temporal resolution, so that spectral changes due to the early relaxation processes following π–π* interband absorption in the pristine polymer could be comprehensively characterized. Absorption occurs in isolated polymer chains in solution and in the solid state (including interchain interactions) for the film. In both cases, we find evidence of delocalization of the electrons and holes formed in the energy bands directly after photoexcitation with excess energy. This is followed by ultrafast (~100 fs) self-localization of the primary photoexcitation and by relatively slow exciton formation (~1 ps). Further relaxation occurs with time constants ranging from hundreds of femtoseconds to tens of picoseconds, due to exciton hopping to sites with lower energy and to a slow conformational planarization of the polymer backbone. Depolarization, a spectral red shift, and important changes in the vibronic structure are observed as a consequence of this relaxation. Finally, relaxed intrachain and interchain singlet excitons are formed in solution and film, respectively, on a 100–200 ps time scale. They decay with a ~500 ps time constant, by intersystem crossing in solution and by nonradiative recombination in the film. Our results are consistent with and strongly support the conclusions we obtained from a similar time-resolved fluorescence study of the polymer PCDTBT (J. Am. Chem. Soc.2010, 132, 17459): ultrafast charge separation in polymer:fullerene blends seems to occur before localization of the primary excitation to form a bound exciton. | ||||||||
 |
|
|||||||
The fluorescence lifetime of the radical cation of N,N,N′,N′-tetramethyl-p-phenylenediamine (Wurster's blue) decreases from 260 ps at 82 K to 200 fs at room temperature. Calculations indicate a small barrier between the excited-state minimum (D1 min) and a conical intersection (CI) of the excited and ground state potentials. The intersection is reached within 200 fs upon torsion of one of the C—N bonds. | ||||||||
|
||||||||
A novel N-substituted 4-methoxy-1,8-naphthalimide (NAFTA 8) especially designed for fluorescent labeling of gold nanoparticles has been synthesized. NAFTA 8 bears a long methylene chain at the imide N atom and has a terminal SH group, which enables its chemical binding to gold nanostructures. The longest wavelength absorption maximum of NAFTA 8 in chloroform is at 370 nm, the fluorescent maximum is at 430 nm and the fluorescent quantum yield is 0.95. The newly synthesized fluorophore is applied for functionalization of gold nanoparticles with diameter 1.5 ± 0.5 nm prepared through chemical reduction. The obtained Monolayer Protected Clusters are characterized by elemental analysis, TEM, XPS, FT-IR, absorption and fluorescence spectroscopy. The performed investigations provide evidence for the formation of chemical bond between the thiol ligand and the gold surface. They also show that the obtained metal/dielectric 3D structures are highly fluorescent. | ||||||||
|
 |
|||||||
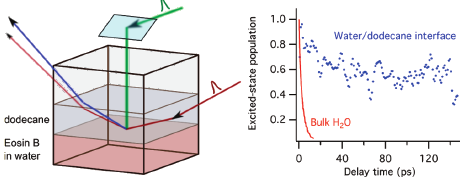
The excited-state dynamics of eosin B (EB) at dodecane/water and decanol/water interfaces has been investigated with polarization-dependent and time-resolved surface second harmonic generation. The results of the polarization-dependent measurements vary substantially with (1) the EB concentration, (2) the age of the sample, and (3) the nature of the organic phase. All of these effects are ascribed to the formation of EB aggregates at the interface. Aggregation also manifests itself in the time-resolved measurements as a substantial shortening of the excited-state lifetime of EB. However, independently of the dye concentration used, the excited-state lifetime of EB at both dodecane/water and decanol/water interfaces is much longer than in bulk water, where the excited-state population undergoes hydrogen-bond-assisted non-radiative deactivation in a few picoseconds. These results indicate that hydrogen bonding between EB and water molecules at liquid/water interfaces is either much less efficient than in bulk water or does not enhance non-radiative deactivation. This strong increase of the excited-state lifetime of EB at liquid/water interfaces opens promising avenues of applying this molecule as a fluorescent interfacial probe. | ||||||||
 |
|
|||||||
The photophysics of two dyes from the xanthene family, eosin B (EB), and eosin Y (EY) has been investigated in various solvents by femtosecond transient absorption spectrosco- py, first, to clarify the huge disparity of the EB fluorescence lifetimes reported in literature, and, second, to understand the mechanism responsible for the ultrafast excited-state deactivation of EB in water. The excited-state lifetime of EB was found to be much shorter in water and in other protic solvents, due to the occurrence of hydrogen-bond assisted nonradiative deactivation. This mechanism is associated with the hydrogen bonds between the solvent molecules and the nitro groups of EB, which become stronger upon optical excitation due to the charge-transfer character of the excited-state. This process is not operative with EY, where the nitro groups are replaced by bromine atoms. Therefore, the excited-state lifetime of EB in solution is directly related to the strength of the solvent as a hydrogen-bond donor, offering the possibility to build a corresponding scale based on the fluorescence quantum yield or lifetime of EB. This scale of hydrogen-bonding strength could be especially useful for studies of liquid interfaces by time-resolved surface second harmonic generation. | ||||||||
|
 |
|||||||
The excited-state dynamics of aminostilbazolium dyes is known to be dominated by nonradiative deactivation through large-amplitude motion. In order to identify the coordinate(s) responsible for this process, the excited-state lifetimes of two dialkylaminostyryl-methylpyridinium iodides have been measured at liquid−liquid interfaces using time-resolved surface second harmonic generation. We found that the decay time of the excited-states of both compounds was increasing with the viscosity of the apolar phase, consisting of n-alkanes of varying length, but was unaffected by that of the polar phase, made of water/glycerol mixtures. This indicates that the nonradiative deactivation is associated with the twist of the dialkylaniline group, which is located in the apolar part of the molecule. | ||||||||
 |
|
|||||||
The photophysics and excited-state dynamics of two dyads consisting of either a free-base or a zinc-tetraphenylporphyrin linked through a rigid bridge to a core-substituted naphthalenediimide (NDI) have been investigated by femtosecond-resolved spectroscopy. The absorption and fluorescence spectra differ substantially from those of the individual units, pointing to a substantial coupling and to a delocalisation of the excitation over the whole molecule, as confirmed by quantum chemistry calculations. A strong dependence of their excited-state dynamics on the solvent polarity has been observed. In toluene, the fluorescence quantum yield of the dyads is of the order of a few percent and the main decay channel of the emitting state is proposed as intersystem-crossing to the triplet state. However, in a medium polarity solvent like dichloromethane, the emitting state undergoes charge separation from the porphyrin to the NDI unit within 1–3 ps, and the ensuing charge-separated state recombines in about 10–20 ps. This solvent dependence can be explained by the weak driving force for charge separation in polar solvents and the large electronic coupling between the porphyrin and NDI moieties, making charge separation a solvent-controlled adiabatic process. | ||||||||
Download this list in a RIS file or a BIB file or a PDF file
Contact:
Eric Vauthey
Physical Chemistry Department - Sciences II - University of Geneva
30, Quai Ernest Ansermet - CH-1211 Geneva 4 (Switzerland)
© All rights reserved by Eric Vauthey and the University of Geneva
Design and code by Guillaume Duvanel