

« Back to all publications
Download this list in a RIS file or a BIB file or a PDF file
|
 |
|||||||
Attached to electron-rich aromatic systems, sulfides are very weak acceptors; however, attached to electron-poor aromatics, they turn into quite strong donors. Here, we show that this underappreciated dual nature of sulfides deserves full consideration for the design of functional systems. Tested with newly designed and synthesized planarizable push−pull mechanophores, sulfide acceptors in the twisted ground state are shown to prevent oxidative degradation and promote blue-shifting deplanarization. Turned on in the planar excited state, sulfide donors promote red-shifting polarization. Impressive Stokes shifts are the result. Demonstrating the usefulness of time-resolved broadband emission spectra to address significant questions, direct experimental evidence for the ultrafast (3.5 ps), polarity-independent and viscosity-dependent planarization from the twisted Franck−Condon S1 state to the relaxed S1 state could be secured. | ||||||||



 |
|
|||||||
The dynamics of a moderately exergonic photoinduced charge separation has been investigated by ultrafast time-resolved infrared absorption with the dimethylanthracene/phthalonitrile donor/acceptor pair in solvents covering a broad range of polarity. A distinct spectral signature of an exciplex could be identified in the −C≡N stretching region. On the basis of quantum chemistry calculations, the 4–5 times larger width of this band compared to those of the ions and of the locally excited donor bands is explained by a dynamic distribution of exciplex geometry with different mutual orientations and distances of the constituents and, thus, with varying charge-transfer character. Although spectrally similar, two types of exciplexes could be distinguished by their dynamics: short-lived, “tight”, exciplexes generated upon static quenching and longer-lived, “loose”, exciplexes formed upon dynamic quenching in parallel with ion pairs. Tight exciplexes were observed in all solvents, except in the least polar diethyl ether where quenching is slower than diffusion. The product distribution of the dynamic quenching depends strongly on the solvent polarity: whereas no significant loose exciplex population could be detected in acetonitrile, both exciplex and ion pair are generated in less polar solvents, with the relative population of exciplex increasing with decreasing solvent polarity. These results are compared with those reported previously with donor/acceptor pairs in different driving force regimes to obtain a comprehensive picture of the role of the exciplexes in bimolecular photoinduced charge separation. | ||||||||

|
 |
|||||||
We report that anion−π and cation−π interactions can occur on the same aromatic surface. Interactions of this type are referred to as ion pair−π interactions. Their existence, nature, and significance are elaborated in the context of spectral tuning, ion binding in solution, and activation of cell-penetrating peptides. The origin of spectral tuning by ion pair−π interactions is unraveled with energy-minimized excited-state structures: The solvent- and pH-independent red shift of absorption and emission of push–pull fluorophores originates from antiparallel ion pair−π attraction to their polarized excited state. In contrast, the complementary parallel ion pair−π repulsion is spectroscopically irrelevant, in part because of charge neutralization by intriguing proton and electron transfers on excited push–pull surfaces. With time-resolved fluorescence measurements, very important differences between antiparallel and parallel ion pair−π interactions are identified and quantitatively dissected from interference by aggregation and ion pair dissociation. Contributions from hydrogen bonding, proton transfer, π–π interactions, chromophore twisting, ion pairing, and self-assembly are systematically addressed and eliminated by concise structural modifications. Ion-exchange studies in solution, activation of cell-penetrating peptides in vesicles, and computational analysis all imply that the situation in the ground state is complementary to spectral tuning in the excited state; i.e., parallel rather than antiparallel ion pair−π interactions are preferred, despite repulsion from the push–pull dipole. The overall quite complete picture of ion pair−π interactions provided by these remarkably coherent yet complex results is expected to attract attention throughout the multiple disciplines of chemistry involved. | ||||||||
 |
|
|||||||
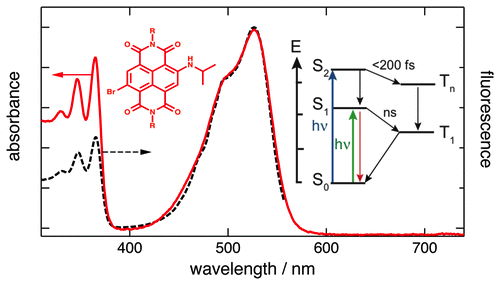
The fluorescence quantum yield of a red naphthalenediimide dye (rNDI) with amino and Br core substituents has been found to decrease by a factor of almost 2 by going from S1 ← S0 to S2 ← S0 excitation. Time-resolved spectroscopic measurements reveal that this deviation from the Kasha−Vavilov’s rule is due to an ultrafast, < 200 fs, intersystem- crossing (ISC) from the S2 state to the triplet manifold, due to the ππ* → nπ* character of the transition and to the presence of the heavy Br atom. In non-core substituted naphthalenediimide (pNDI), ISC is slower, ∼2 ps, and was found to be reversible on a time scale shorter than that of vibrational cooling. The fluorescence and triplet quantum yields of rNDI, thus, can be substantially changed by a simple variation of the excitation wavelength. | ||||||||
|
 |
|||||||
The femto- to microsecond excited-state dynamics of an electron donor–acceptor–donor triad, consisting of two red core-substituted naphthalenediimides (rNDI) and one colorless naphthalenediimide (pNDI), in solution has been compared to that of a supramolecular surface architecture, constituted of coaxial stacks of rNDI and pNDI and prepared by self-organizing surface initiated polymerization (SOSIP). In the triad, charge separation between an excited rNDI and pNDI takes place in highly polar solvents only and for a subensemble of molecules, around 30%, with a folded conformation. Other processes, such as singlet and triplet excitation energy transfer from pNDI to rNDI and intersystem crossing, are also operative. Additionally, bimolecular symmetry-breaking charge separation upon triplet–triplet annihilation is observed on the microsecond time scale in polar solvent. In the surface architecture, excitation of an rNDI is followed by an ultrafast symmetry breaking-charge separation resulting in a charge-transfer exciton, which either recombines or dissociates into a charge-separated state with the electron and the hole in different stacks. The same charge-separated state can also be populated upon excitation of pNDI, either via a charge-transfer pNDI exciton or after excitation energy transfer to rNDI. Charge recombination in the SOSIP film takes place on a wide range of time scales, ranging from a few picoseconds to several hundreds of microseconds. | ||||||||
 |
|
|||||||
Two donor bridge–acceptor molecules with terminal triarylamine and Ru(bpy)32+ (bpy = 2,2′-bipyridine) redox partners were synthesized and investigated by cyclic voltammetry, optical absorption, luminescence, and transient absorption spectroscopy. The two dyads differ only by the central bridging unit, which was tetramethoxybenzene (tmb) in one case and unsubstituted phenylene (ph) in the other case. Photoirradiation of the Ru(bpy)32+ complex of the two dyads triggers intramolecular electron transfer from the triarylamine to the 3MLCT-excited metal complex, and this process occurs with time constants of 1.5 and 6.8 ns for the tmb- and ph-bridged dyads, respectively. Thermal electron transfer in the reverse direction then leads to disappearance of the photoproduct with a time constant of 10 ns in both dyads. The faster rate of photoinduced charge transfer in the tmb-bridged dyad can be understood in the framework of a hole-tunneling model in which the electron-rich tmb bridge imposes a more shallow barrier than the less electron-rich ph spacer. Until now tmb-based molecular wires have received very little attention, and alkoxy substituents have been mostly used for improving the solubility of oligo-p-phenylene vinylene (OPV) and oligo-p-phenylene ethynylene (OPE) wires. Our study illustrates how four alkoxy-substituents on a phenylene backbone can have a significant influence on the charge-transfer properties of a molecular wire, and this is relevant in the greater context of a future molecular electronics technology. | ||||||||
|
 |
|||||||
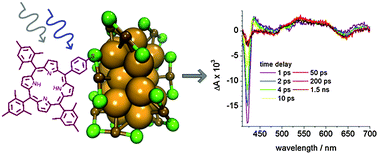
A protected S-acetylthio porphyrin was synthesized and attached to the Au38(2-phenylethanethiolate)24 cluster in a ligand exchange reaction. Chiral high performance liquid chromatography of the functionalized cluster yielded enantiomeric pairs of clusters probably differing in the binding site of the porphyrin. As proven by circular dichroism, the chirality was maintained. Exciton coupling between the cluster and the chromophore is observed. Zinc can be incorporated into the porphyrin attached to the cluster, as evidence by absorption and fluorescence spectroscopy, however, the reaction is slow.Quenching of the chromophores fluorescence is observed, which can be explained by energy transfer from the porphyrin to the cluster. Transient absorption spectra on the Au38(2-phenylethanethiolate)24 and the functionalized cluster probe the bleach of the gold cluster due to ground state absorption and characteristic excited state absorption signals. Zinc incorporation does not have a pronounced effect on the photophysical behaviour. Decay times are typical for the molecular behaviour of small monolayer protected gold clusters. | ||||||||
 |
|
|||||||
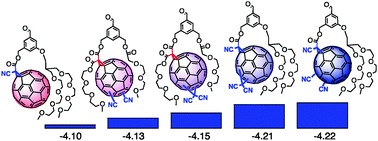
Fullerenes of increasing electron deficiency are designed, synthesized and evaluated in multicomponent surface architectures to ultimately build gradients in LUMO levels with nine components over 350 meV down to −4.22 eV. | ||||||||
|
 |
|||||||
In this report, “fluorescent flippers” are introduced to create planarizable push–pull probes with the mechanosensitivity and fluorescence lifetime needed for practical use in biology. Twisted push–pull scaffolds with large and bright dithienothiophenes and their S,S-dioxides as the first “fluorescent flippers” are shown to report on the lateral organization of lipid bilayers with quantum yields above 80% and lifetimes above 4 ns. Their planarization in liquid-ordered (Lo) and solid-ordered (So) membranes results in red shifts in excitation of up to +80 nm that can be transcribed into red shifts in emission of up to +140 nm by Förster resonance energy transfer (FRET). These unique properties are compatible with multidomain imaging in giant unilamellar vesicles (GUVs) and cells by confocal laser scanning or fluorescence lifetime imaging microscopy. Controls indicate that strong push–pull macrodipoles are important, operational probes do not relocate in response to lateral membrane reorganization, and two flippers are indeed needed to “really swim,” i.e., achieve high mechanosensitivity. | ||||||||
 |
|
|||||||
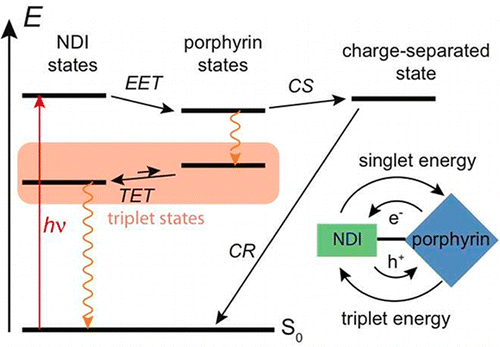
The excited-state dynamics of two molecular dyads, consisting of zinc (1) and free-base (2) porphyrin connected via a peptide linker to a core-substituted naphthalenediimide (NDI) have been investigated using optical spectroscopy. These dyads exhibit rich photophysics because of the large number of electronic excited states below 3 eV. In the case of 1 in apolar solvents, excitation energy transfer from the vibrationally hot singlet excited porphyrin to the NDI takes place with a 500 fs time constant. Electronic energy ends up in the NDI-localized triplet state, which decays to the ground state on a microsecond timescale. In polar solvents, ground-state recovery is faster by 5 orders of magnitude because of the occurrence of charge separation followed by recombination. On the other hand, excitation energy transfer in 2 takes place in the opposite direction, namely from the NDI to the porphyrin, which then undergoes intersystem crossing to the triplet state, followed by triplet energy transfer back to the NDI. Therefore, four distinct local electronic excited states are consecutively populated after excitation of the NDI unit of 2, with the energy shuttling between the two ends of the dyad. | ||||||||
|
 |
|||||||
Differently substituted anils (Schiff bases) and their boranil counterparts lacking the proton-transfer functionality have been studied using stationary and femtosecond time-resolved absorption, fluorescence, and IR techniques, combined with quantum mechanical modelling. Dual fluorescence observed in anils was attributed to excited state intramolecular proton transfer. The rate of this process varies upon changing solvent polarity. In the nitro-substituted anil, proton translocation is accompanied by intramolecular electron transfer coupled with twisting of the nitrophenyl group. The same type of structure is responsible for the emission of the corresponding boranil. A general model was proposed to explain different photophysical responses to different substitution patterns in anils and boranils. It is based on the analysis of changes in the lengths of CN and CC bonds linking the phenyl moieties. The model allows predicting the contributions of different channels that involve torsional dynamics to excited state depopulation. | ||||||||
 |
|
|||||||
The electronic absorption spectrum of 3-hydroxyflavone (3HF) in various solvents exhibits a long-wavelength (LW) band, whose origin has been debated. The excited-state dynamics of neutral and basic solutions of 3HF in alcohols upon excitation in this LW band has been investigated using a combination of fluorescence up-conversion and transient electronic and vibrational absorption spectroscopies. The ensemble of results reveals that, in neutral solutions, LW excitation results in the population of two excited species with similar fluorescence spectra but very different lifetimes, namely 40–100 ps and 2–3 ns, depending on the solvent. In basic solutions, the relative concentrations of these species change considerably in favor of that with the short-lived excited state. On the basis of the spectroscopic data and quantum chemistry calculations, the short lifetime is attributed to the excited state of 3HF anion, whereas the long one is tentatively assigned to an excited hydrogen-bonded complex with the solvent. Excited-state intermolecular proton transfer from the solvent to the anion yielding the tautomeric form of 3HF is not operative, as the excited anion decays to the ground state via an efficient nonradiative transition. | ||||||||
|
||||||||
The role of ligand-field states for the photophysical properties of d6 systems has been discussed in a large number of publications over the past decades. Since the seminal paper by Houten and Watts, for instance, the quenching of the 3MLCT luminescence in ruthenium(II) polypyridyl complexes is attributed to the presence of the first excited ligand-field state, namely a component of the 3T1(t2g5eg1) state, at similar energies. If this state lies above the 3MLCT state, the luminescence is quenched via thermal population at elevated temperatures only. If it lies well below, then the luminescence is quenched down to cryogenic temperatures. In this contribution we present transient absorption spectra on non-luminescent ruthenium polypyridyl complexes such as [Ru(m-bpy)3]2+, m-bpy = 6-methyl-2,2’-bipyridine, in acetonitrile at room temperature, which reveal an ultra-rapid depopulation of the 3MLCT state but a much slower ground state recovery. We propose that in this and related complexes the methyl groups force longer metal-ligand bond lengths, thus resulting in a lowering of the ligand-field strength such that the 3dd state drops to below the 3MLCT state, and that furthermore the population of this state from the 3MLCT state occurs faster than its decay to the ground state. In addition we demonstrate that in this complex the luminescence can be switched on by external pressure, which we attribute to a destabilisation of the ligand-field state by the pressure due to its larger molecular volume compared to the ground state as well as the 3MLCT state. | ||||||||
Download this list in a RIS file or a BIB file or a PDF file
Contact:
Eric Vauthey
Physical Chemistry Department - Sciences II - University of Geneva
30, Quai Ernest Ansermet - CH-1211 Geneva 4 (Switzerland)
© All rights reserved by Eric Vauthey and the University of Geneva
Design and code by Guillaume Duvanel