

« Back to all publications
Download this list in a RIS file or a BIB file or a PDF file
|
 |
|||||||
Intermolecular H‐bonding dynamics around a photoexcited quadrupolar dye is directly observed using transient 2D‐IR spectroscopy. Upon solvent‐induced symmetry breaking, the H‐bond accepting abilities of the two nitrile end‐groups change drastically, and in extremely protic (superprotic) solvents, a tight H‐bond complex forms at one end. The time evolution of the 2D C≡N lineshape in methanol points to rapid, 2–3 ps, spectral diffusion due to fluctuations of the H‐bonding network. Similar behavior is observed in a superprotic solvent shortly after photoexcitation of the dye. However, at later times, the completely inhomogeneous band does not exhibit spectral diffusion for at least 5 ps, pointing to a glass‐like environment around one side of the dye. About half of the excited dyes show this behavior attributed to the tight H‐bond complex, whereas the others are loosely bound. A weak cross peak indicates partial exchange between these excited state subpopulations. | ||||||||



 |
|
|||||||
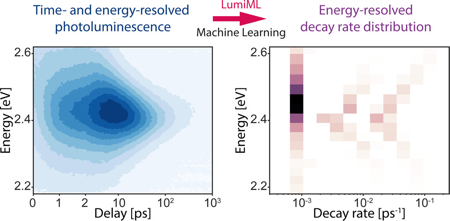
Time-resolved photoluminescence is one of the most standard techniques to understand and systematically optimize the performance of optical materials and optoelectronic devices. Here, we present a machine learning code to analyze time-resolved photoluminescence data and determine the decay rate distribution of an arbitrary emitter without any a priori assumptions. To demonstrate and validate our approach, we analyze computer-generated time-resolved photoluminescence data sets and show its benefits for studying the photoluminescence of novel semiconductor nanocrystals (quantum dots), where it quickly provides insight into the possible physical mechanisms of luminescence without the need for educated guessing and fitting. | ||||||||
|
 |
|||||||
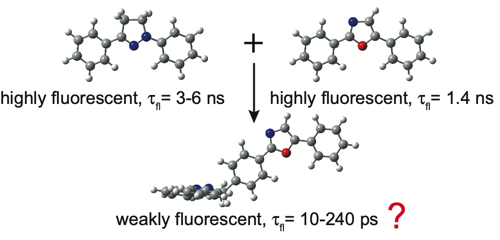
The excited-state dynamics of a T-shaped bichromophoric molecule, consisting of two strong fluorophores, diphenyloxazole and diphenylpyrazoline, directly linked in an orthogonal geometry, was investigated. Despite the weak coupling ensured by this geometry and confirmed by the electronic absorption spectra, this dyad exhibits only weak fluorescence in both apolar and polar solvents, with fluorescence lifetimes ranging from 200 ps in CHX to 10 ps in ACN. Ultrafast spectroscopic measurements reveal that the fluorescence quenching in polar solvents is due to the population of a charge-separated state. In non-polar solvents, this process is energetically not feasible, and a quenching due to an efficient intersystem crossing (ISC) to the triplet manifold is proposed, based on quantum-chemical calculations. This process occurs via the spin–orbit charge-transfer (SOCT) ISC mechanism, which is enabled by the charge-transfer character acquired by the S1 state of the dyad upon structural relaxation and by the orthogonal arrangement of the molecular orbitals involved in the transition. The same mechanism is proposed to explain why the recombination of the charge-separated state is faster in medium than in highly polar solvents, as well as to account for the fast decay of the lowest triplet state to the ground state. | ||||||||
 |
|
|||||||
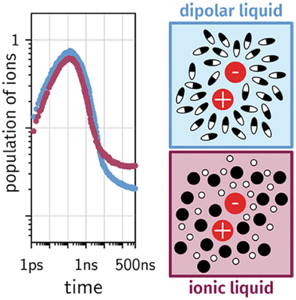
Bimolecular photoinduced electron transfer between perylene and two quenchers was investigated in an imidazolium room-temperature ionic liquid (RTIL) and in a dipolar solvent mixture of the same viscosity using transient absorption on the subpicosecond to submicrosecond time scales. Whereas charge separation dynamics were similar in both solvents, significant differences were observed in the temporal evolution of the ensuing radical ions: although small, the free-ion yield is significantly larger in the RTIL, and recombination of the ion pair to the triplet state of perylene is more efficient in the dipolar solvent. The temporal evolution of reactant, ion, and triplet state populations could be well reproduced using unified encounter theory. This analysis reveals that the observed differences can be explained by the strong screening of the Coulomb potential in the ion pair by the ionic solvent. In essence, RTILs favor free ions compared to highly dipolar solvents of the same viscosity. | ||||||||
|
||||||||
Two-photon induced polymerization (2PP) based 3D printing is a powerful microfabrication tool. Specialized two-photon initiators (2PIs) are critical components of the employed photosensitive polymerizable formulations. This work investigates the cooperative enhancement of two-photon absorption cross sections (σ2PA) in a series of 1,3,5-triazine-derivatives bearing 1-3 aminostyryl-donor arms, creating dipolar, quadrupolar and octupolar push-pull systems. The multipolar 2PIs were successfully prepared and characterized, σ2PA were determined using z-scan at 800 nm as well as spectrally resolved two-photon excited fluorescence measurements, and the results were compared to high-level ab initio computations. Modern tunable femtosecond lasers allow 2PP-processing at optimum wavelengths tailored to the absorption behavior of the 2PI. 2PP structuring tests revealed that while performance at 800 nm is similar, at their respective σ2PA-maxima the octupolar triazine-derivative outperforms a well-established ketone-based quadrupolar reference 2PI, with significantly lower fabrication threshold at exceedingly high writing speeds up to 200 mm/s and a broader window for ideal processing parameters. | ||||||||
|
||||||||
Previously, we have shown that the use of a cyclopeptidic carrier could be of great interest for the design of fully characterized prodrugs for further use in photodynamic therapy. In order to further optimize the design, we decided to modify the highly quenched conjugate uPA-cPPP4/5 by co-loading a long-distance fluorescence quencher. For this purpose we tethered two black hole quenchers (BHQ3) together with two pheophorbide A moities onto the same PEGylated backbone and assessed the modified photophysical properties. In addition, to prove the reliability of our concept, we designed two analogues, uPA-cPPQ2+2/5 and CathB-cPPQ2+2/5, by using two different peptidic linkers as substrates for uPA and cathepsin B, respectively. These two conjugates proved to be much more water-soluble than their analogues bearing only Phas. These conjugates are not only highly quenched in their native state with regard to their fluorescence emission (up to 850 ± 287 times less fluorescent for CathB-cPPQ2+2/5 as compared to the unquenched monosubstituted reference uPA-cPPP1/5), but also prevent singlet oxygen production (with a total quenching of the emission when the quenchers are co-loaded with photosensitizers) when the photosentistizers are excited. After proteolytic activation, these conjugates recover their photophysical properties in the same way as occurred for uPA-cPPP4/5, with up to a 120-fold increase in fluorescence emission for uPA-cPPQ2+2/5 after two hours of incubation with uPA. | ||||||||
|
||||||||
Herein, we report the synthesis of a new prodrug system consisting of regioselectively addressable functionalized templates bearing multiple pheophorbide A moieties for use in photodynamic therapy. These coupling reactions were achieved using copper-free “click” chemistry, namely a strain-promoted azide–alkyne cycloaddition. This new design was used to obtain well-defined quenched photosensitizer prodrugs with perfect knowledge of the number and position of loaded photosensitizers, providing structures bearing up to six photosentitizers and two PEG chains. These conjugates are ideally quenched in their native state regarding their fluorescence emission (up to 155 ± 28 times less fluorescent for an hexasubstituted conjugate than a monosubstituted non-quenched reference compound) or singlet oxygen production (decreased 8.7-fold in the best case) when excited. After 2 h of proteolytic activation, the fluorescence emission of a tetrasubstituted conjugate was increased 17-fold compared with the initial fluorescence emission. | ||||||||
|
||||||||
The ultimate goal of chemical kinetics is to understand why a given reaction is fast or not. To this end it is necessary to count on robust and experimentally well tested theories. One of the difficulties, long recognized in the study of bimolecular reactions, is the role of the molecular displacement, i.e. diffusion. Nonetheless the field is still lacking a compelling amount of case studies contrasting physical models to experiments. By performing transient absorption experiments on the photo-induced electron transfer reaction between perylene and N,N-dimethylaniline in liquid solutions over many orders of magnitude in time, we try to understand the factors determining the kinetics and yields of the full photocycle. We present a method to overcome potential pitfalls in the extraction of the relevant quantities, the transient populations, from the experimental data due to the changes in band shapes and positions. The results are compared to simulations of two different theories: a reaction–diffusion approach based on the encounter theories, and a formal kinetic scheme. We conclude that while the former explains the observed trends in the kinetics with quencher concentration and viscosity exceptionally well, the latter fails. Moreover the analysis of the data with the assistance of encounter theory unveils effects that otherwise would pass unnoticed. This approach and its results exemplify the path to follow in other condensed media whenever diffusion is involved. | ||||||||
|
||||||||
Bent N,N′‐diphenyl‐dihydrodibenzo[a,c]phenazine amphiphiles are introduced as mechanosensitive membrane probes that operate by an unprecedented mechanism, namely, unbending in the excited state as opposed to the previously reported untwisting in the ground and twisting in the excited state. Their dual emission from bent or “closed” and planarized or “open” excited states is shown to discriminate between micelles in water and monomers in solid‐ordered (So), liquid‐disordered (Ld) and bulk membranes. The dual‐emission spectra cover enough of the visible range to produce vesicles that emit white light with ratiometrically encoded information. Strategies to improve the bent mechanophores with expanded π systems and auxochromes are reported, and compatibility with imaging of membrane domains in giant unilamellar vesicles by two‐photon excitation fluorescence (TPEF) microscopy is demonstrated. | ||||||||
|
 |
|||||||
Diketopyrrolopyrroles (DPPs) have recently attracted much interest as very bright and photostable red‐emitting molecules. However, their tendency to form nonfluorescent aggregates in water through the aggregation‐caused quenching (ACQ) effect is a major issue that limits their application under the microscope. Herein, two DPP molecules have been incorporated into the membrane of highly stable and water‐soluble quatsomes (QS; nanovesicles composed of surfactants and sterols), which allow their nanostructuration in water and, at the same time, limits the ACQ effect. The obtained fluorescent organic nanoparticles showed superior structural homogeneity, along with long‐term colloidal and optical stability. A thorough one‐ (1P) and two‐photon (2P) fluorescence characterization revealed the promising photophysical features of these fluorescent nanovesicles, which showed a high 1P and 2P brightness. Finally, the fluorescent QSs were used for the in vitro bioimaging of Saos‐2 osteosarcoma cell lines; this demonstrates their potential as nanomaterials for bioimaging applications. | ||||||||
|
||||||||
Kynurenines (KNs) are natural UV filters of the human eye lens, protecting the eye tissues from solar UV radiation. Key points of their effective protection are the intramolecular charge transfer (ICT) in the excited state and the fast dissipation of absorbed light energy into heat via the intermolecular H-bonds. Herein we report that the introduction of an unsaturated double bond in the amino acid side chain, operating as an ICT-enhancing electron donor group, drastically accelerates the internal conversion (IC) due to a conical intersection (CI) between the potential energy surfaces of the excited and ground states. Our photophysical study of a deaminated KN (carboxyketoalkene, CKA), an intrinsic product of KN thermal decomposition, demonstrates an unusually fast excited state decay in a broad range of solvents of different polarity and proticity. The detailed analysis of interactions in the excited state by different computational techniques revealed that the changes in molecular structure – the twist of the carbonyl group from the plane of the aromatic ring followed by the formation of two mutually orthogonal conjugated substructures – are responsible for the CI of the excited and ground state potential energy surfaces. Intermolecular H-bonds facilitate the transition to the CI, but do not play a crucial role in the fast decay of the excited state. An extremely fast and efficient IC in CKA opens the way for the design of new types of organic UV filters and their applications in material science, cosmetics and medicine. | ||||||||
 |
|
|||||||
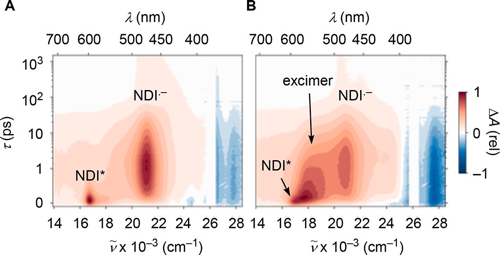
In this report, we demonstrate that synergistic effects between π–π stacking and anion−π interactions in π-stacked foldamers provide access to unprecedented catalytic activity. To elaborate on anion–(π)n–π catalysis, we have designed, synthesized and evaluated a series of novel covalent oligomers with up to four face-to-face stacked naphthalenediimides (NDIs). NMR analysis including DOSY confirms folding into π stacks, cyclic voltammetry, steady-state and transient absorption spectroscopy the electronic communication within the π stacks. Catalytic activity, assessed by chemoselective catalysis of the intrinsically disfavored but biologically relevant addition reaction of malonate half thioesters to enolate acceptors, increases linearly with the length of the stacks to reach values that are otherwise beyond reach. This linear increase violates the sublinear power laws of oligomer chemistry. The comparison of catalytic activity with ratiometric changes in absorption and decreasing energy of the LUMO thus results in superlinearity, that is synergistic amplification of anion−π catalysis by remote control over the entire stack. In computational models, increasing length of the π-stacked foldamers correlates sublinearly with changes in surface potentials, chloride binding energies, and the distances between chloride and π surface and within the π stack. Computational evidence is presented that the selective acceleration of disfavored but relevant enolate chemistry by anion−π catalysis indeed originates from the discrimination of planar and bent tautomers with delocalized and localized charges, respectively, on π-acidic surfaces. Computed binding energies of keto and enol intermediates of the addition reaction as well as their difference increase with increasing length of the π stack and thus reflect experimental trends correctly. These results demonstrate that anion–(π)n–π interactions exist and matter, ready for use as a unique new tool in catalysis and beyond. | ||||||||
|
 |
|||||||
A planarizable push–pull molecular probe with mechanosensitive properties was investigated at several biomimetic interfaces, consisting of different phospholipid monolayers located between dodecane and an aqueous buffer solution, using the interface-specific surface-second-harmonic-generation (SSHG) technique. Whereas the SSHG spectra recorded at liquid-disordered interfaces were similar to the absorption spectra in bulk solutions, those measured at liquid-ordered phases exhibited a remarkable shift towards lower energies to an extent depending on the surface pressure of the phospholipid monolayer. On the basis of quantum-chemical calculations, this effect was accounted for by the planarization of the mechanosensitive probe. Polarization-resolved SSHG measurements revealed that the average orientation of the probe at the interface is an even more sensitive reporter of lateral pressure and order than the spectral shape. Additionally, time-resolved SSHG measurements pointed to slower dynamics upon intercalation inside the phospholipid monolayer, most likely due to the more constrained environment. This study demonstrates that the concept of mechanosensitive optical probes can be further exploited when combined with a surface-selective nonlinear optical technique. | ||||||||
|
||||||||
The enzyme catechol-O-methyltransferase (COMT) has water soluble (S-COMT) and membrane associated (MB-COMT), bitopic, isoforms. Of these MB-COMT is a drug target in relation to the treatment of Parkinson's disease. Using a combination of computational and experimental protocols, we have determined the substrate selection mechanism specific to MB-COMT. We show: (1) substrates with preferred affinity for MB-COMT over S-COMT orient in the membrane in a fashion conducive to catalysis from the membrane surface and (2) binding of COMT to its cofactor ADOMET induces conformational change that drives the catalytic surface of the protein to the membrane surface, where the substrates and Mg2+ ions, required for catalysis, are found. Bioinformatics analysis reveals evidence of this mechanism in other proteins, including several existing drug targets. The development of new COMT inhibitors with preferential affinity for MB-COMT over S-COMT is now possible and insight of broader relevance, into the function of bitopic enzymes, is provided. | ||||||||
 |
|
|||||||
The applicability of room-temperature ionic liquids (RTILs) as inert solvents is generally based on their electrochemical window. We herein show that this concept has its limitations if RTILs are exposed to an oxidizing environment in the presence of light. Acetonitrile solutions of RTILs with 1-methyl-3-ethylimidazolium as cation and five different anions, including thiocyanate (SCN–) and dicyanamide (DCA–), were investigated. Upon addition of organic electron acceptors to solutions of RTILs with SCN– or DCA–, charge-transfer (CT) absorption bands due to the formation of donor–acceptor complexes between the anion and the electron acceptor were observed. Time-resolved measurements from the femtosecond to the microsecond regimes were used to investigate the nature and the excited-state dynamics of these complexes upon excitation in the CT band. We show that even though the RTILs are seemingly inert according to their electrochemical properties, the dicyanamide and thiocyanate based RTILs can actively participate in photochemical reactions in oxidizing environments and therefore differ from the behavior expected for an inert solvent. This has not only important implications for the long-term stability of RTIL-based systems but can also lead to misinterpretation of photochemical studies in these solvents. | ||||||||
|
 |
|||||||
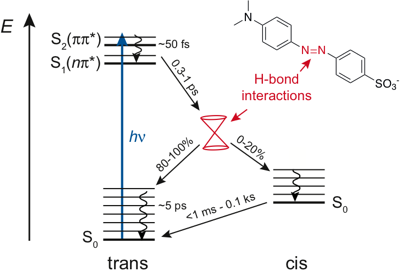
The excited-state dynamics of the push–pull azobenzene Methyl Orange (MO) were investigated in several solvents and water/glycerol mixtures using a combination of ultrafast time-resolved fluorescence and transient absorption in both the UV-visible and the IR regions, as well as quantum chemical calculations. Optical excitation of MO in its trans form results in the population of the S2 ππ* state and is followed by internal conversion to the S1 nπ* state in ∼50 fs. The population of this state decays on the sub-picosecond timescale by both internal conversion to the trans ground state and isomerisation to the cis ground state. Finally, the cis form converts thermally to the trans form on a timescale ranging from less than 50 ms to several minutes. Significant differences depending on the hydrogen-bond donor strength of the solvents, quantified by the Kamlet Taft parameter α, were observed: compared to the other solvents, in highly protic solvents (α > 1), (i) the viscosity dependence of the S1 state lifetime is less pronounced, (ii) the S1 state lifetime is shorter by a factor of ≈1.5 for the same viscosity, (iii) the trans-to-cis photoisomerisation efficiency is smaller, and (iv) the thermal cis-to-trans isomerisation is faster by a factor of ≥103. These differences are explained in terms of hydrogen-bond interactions between the solvent and the azo nitrogen atoms of MO, which not only change the nature of the S1 state but also have an impact on the shape of ground- and excited-state potentials, and, thus, affect the deactivation pathways from the excited state. | ||||||||
|
||||||||
A series of bis(pyreneamide) macrocycles, synthesized in two steps from THF, THP, oxepane and 1,4-dioxane, are tested as chemosensors for a large range of mono-, di- and trivalent cations. In their native states, these macrocycles exhibit a strong excimer fluorescence that is quenched upon the addition of the metal ions (alkaline, alkaline earth, p-, d-, and f-block metals). UV-Vis spectrophotometric titrations, cyclic voltammetry, excimer fluorescence quenching and transient absorption spectroscopy experiments helped characterize the On-Off changes occurring upon binding and demonstrate that the highest stability constants are obtained with divalent cations Ca2+ and Ba2+ specifically. | ||||||||
|
||||||||
The photophysical properties of two pyrene-bodipy molecular dyads, composed of a phenyl-pyrene (Py-Ph) linked to the meso position of a bodipy (BD) molecule with either H-atoms (BD1) or ethyl groups (BD2) at the 2, 6 positions, are investigated by stationary, nanosecond and femtosecond spectroscopy. The properties of these dyads (Py-Ph-BD1 and Py-Ph-BD2) are compared to those of their constituent chromophores in two solvents namely 1,2 dichloroethane (DCE) and acetonitrile (ACN). Stationary spectroscopy reveals a weak coupling among the subunits in both dyads. Excitation of the Py subunit eads to emission that is totally governed by the BD subunits in both dyads pointing to excitation energy transfer (EET) from the Py to BD chromophore. Femtosecond fluorescence and transient absorption spectroscopy reveal that EET takes place within 0.3-0.5 ps and is mostly independent of the solvent and the type of the BD subunit. The EET lifetime is in reasonable agreement with that predicted by Förster theory. After EET has taken place, Py-Ph-BD1 in DCE and Py-Ph-BD2 in both solvents decay mainly radiatively to the ground state with 3.5 - 5.0 ns lifetimes which are similar to those of the individual BD chromophores. However, the excited state of Py-Ph-BD1 in ACN is quenched having a lifetime of 1 ns. This points to the opening of an additional non-radiative channel of the excited state of Py-Ph-BD1 in this solvent, most probably charge separation (CS). Target analysis of the TA spectra has shown that the CS follows an inverted kinetics and is substantially slower than the recombination of the charge-separated state. Occurrence of CS with Py-Ph-BD1 in ACN is also supported by energetic considerations. The above results indicate that only a small change in the structure of the BD units incorporated in the dyads, significantly affects the excited state dynamics leading either to a dyad with long lifetime and high fluorescence quantum yield or to a dyad with an intramolecular CS ability. | ||||||||
 |
|
|||||||
Azetidinyl substituents have been recently used to improve the fluorescence quantum yield of several classes of fluorophores. Herein, we demonstrate that other useful photochemical processes can be modulated using this strategy. In particular, we prepared and measured the quantum yield of photorelease of a series of 7-azetidinyl-4-methyl coumarin esters and compared it to their 7-diethylamino and julolidine-fused analogues. The efficiency of the photorelease reactions of the azetidinyl-substituted compounds was 2- to 5-fold higher than the corresponding diethylamino coumarins. We investigated the origin of this effect in model fluorophores and in the photoactivatable esters, and found that H-bonding with the solvent seems to be the prominent deactivation channel inhibited upon substitution with an azetidinyl ring. We anticipate that this substitution strategy could be used to modulate other photochemical processes with applications in chemical biology, catalysis and materials science. | ||||||||
Download this list in a RIS file or a BIB file or a PDF file
Contact:
Eric Vauthey
Physical Chemistry Department - Sciences II - University of Geneva
30, Quai Ernest Ansermet - CH-1211 Geneva 4 (Switzerland)
© All rights reserved by Eric Vauthey and the University of Geneva
Design and code by Guillaume Duvanel