

« Back to all publications
Download this list in a RIS file or a BIB file or a PDF file
|
 |
|||||||
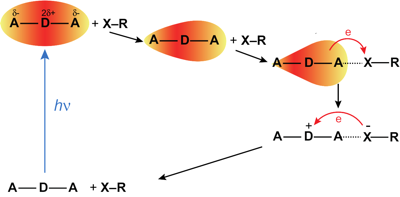
The formation of a halogen-bond (XB) complex in the excited state was recently reported with a quadrupolar acceptor–donor–acceptor dye in two iodine-based liquids (J. Phys. Chem. Lett. 2017, 8, 3927–3932). The ultrafast decay of this excited complex to the ground state was ascribed to an electron transfer quenching by the XB donors. We examined the mechanism of this process by investigating the quenching dynamics of the dye in the S1 state using the same two iodo-compounds diluted in inert solvents. The results were compared with those obtained with a non-halogenated electron acceptor, fumaronitrile. Whereas quenching by fumaronitrile was found to be diffusion controlled, that by the two XB compounds is slower, despite a larger driving force for electron transfer. A Smoluchowski–Collins–Kimball analysis of the excited-state population decays reveals that both the intrinsic quenching rate constant and the quenching radius are significantly smaller with the XB compounds. These results point to much stronger orientational constraint for quenching with the XB compounds, indicating that electron transfer occurs upon formation of the halogen bond. | ||||||||



 |
|
|||||||
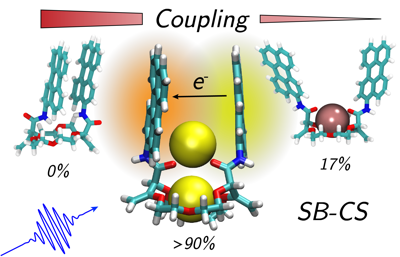
Understanding structure–property relationships in multichromophoric molecular architectures is a crucial step in establishing new design principles in organic electronics as well as to fully understand how nature exploits solar energy. Here, we study the excited state dynamics of three bichromophores consisting of two perylene chromophores linked to three different crown-ether backbones, using stationary and ultrafast electronic spectroscopy combined with molecular dynamics simulations. The conformational space available to the bichromophores depends on the structure and geometry of the crown-ether and can be significantly changed upon cation binding, strongly affecting the excited-state dynamics. We show that, depending on the conformational restrictions and the local environment, the nature of the excited state varies greatly, going from an excimer to a symmetry-broken charge separated state. These results can be rationalised in terms of a structure–property relationship that includes the effect of the local environment. | ||||||||
|
 |
|||||||
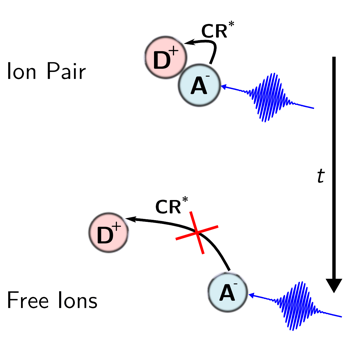
The excited-state dynamics of the radical anion of perylene (Pe) generated upon bimolecular photoinduced electron transfer (PET) with a donor was investigated using broadband pump–pump–probe spectroscopy. It was found to depend on the age of the anion, that is, on the time interval between the first pump pulse that triggers PET and the second one that excites the ensuing Pe anion (Pe•–). These differences, observed in acetonitrile but not in tetrahydrofuran, report on the evolution of the PET product from an ion pair to free ions. Two photoinduced charge recombination pathways of the ion pair to the neutral Pe*(S1) + donor state were identified: one occurring in a few picoseconds from Pe•–*(D1) and one taking place within 100–200 fs from Pe•–*(Dn>1). Both processes are sensitive to the interionic distance over different length scales and thus serve as molecular rulers. | ||||||||
|
 |
|
|||||||
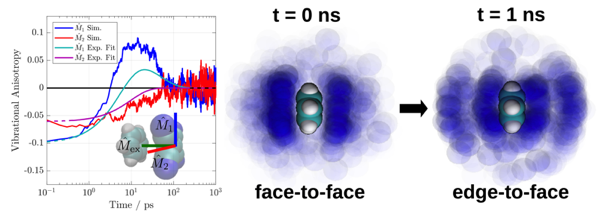
The structural dynamics of an electron donor/acceptor complex (DAC) consisting of benzene and tetracyanoethylene (Bz/TCNE) solvated in CH2Cl2 have been investigated using ultrafast spectroscopy and mixed quantum/classical computer simulations. Population dynamics from visible and infrared transient absorption (TRIR) spectroscopy point to complex sub-10 ps dynamics followed by charge recombination on a 55 to 60 ps timescale. Structural dynamics involving large-scale reorganization of radical ion pairs are revealed using TRIR anisotropy measurements. A computational study combining quantum chemical calculations and classical molecular dynamics simulations was able to reproduce the experimental electronic absorption lineshape and TRIR anisotropy dynamics, allowing for a detailed investigation of the pair conformational dynamics. Contrary to the static single structure typically assumed in descriptions of DACs, we find that neither the ground nor excited state can be described using a single, well-defined species. Instead, the pair undergoes a rearrangement from disordered pi-stacks to edge-to-face T-shaped structures following excitation. Translational diffusion of the radical ion pairs following excitation was found to be heterogeneous and dependent on both pair separation and orientation coordinates. Given the sensitivity of charge-transfer reactions to the arrangement of donor/acceptor pairs, the structural heterogeneity and corresponding dynamics demonstrated herein must be taken into account in future modeling of charge recombination processes in DACs. | ||||||||
|
 |
|||||||
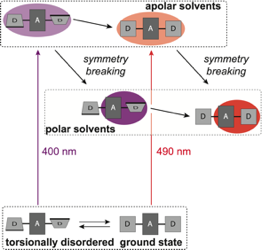
The influence of torsional disorder around the ethynyl pi-bridges of a linear D-pi-A-pi-D molecule on the nature of its S1 excited state was investigated using ultrafast time-resolved infrared spectroscopy. By tuning the pump wavelength throughout the S1<- S0 absorption band, subpopulations with different extents of asymmetry could be excited. In non-polar solvents, the equilibrated S1 state is symmetric and quadrupolar independently of the initial degree of distortion. Photoexcitation of distorted molecules is followed by planarization and symmetrization of the S1 state. Excited-state symmetry breaking is only observed in polar environments, where the equilibrated S1 state has a strong dipolar character. However, neither the extent nor the rate of symmetry breaking are enhanced in an initially distorted molecule. They are only determined by the polarity and the dynamic properties of the solvent. | ||||||||
|
||||||||
In this work we present the design, synthesis and systematic investigation of the optical properties of symmetric triphenylamine (TPA)-substituted thiophenes. The use of electron-donating (-OMe, -tBu, -Me, -TMS), -neutral (-H) or -withdrawing (-F, -CN, -SO2Me) substituents gives rise to D-A-D based two-photon absorption (2PA) chromophores. The photophysical properties of these compounds, including one-photon absorption and 2PA using two-photon-excited fluorescence, were investigated in different organic solvents with varying polarity. The maximum 2PA cross sections prove to be strongly dependent on the nature of the TPA substituent and range between ca. 173 GM (Goeppert-Mayer units) and 379 GM. Although most of the investigated substances also exhibit high fluorescence quantum yields, two-photon absorption screening tests of an acrylate monomer formulation revealed the efficiency of these materials as 2PA photoinitiators. These results are supported by quantum chemical calculations of the spin density distribution indicating that the mechanism of polymerization initiation using acrylate monomer is favored by strong localization of the unpaired electrons in the triplet state on the C2 carbon of the thiophene moiety. | ||||||||
 |
|
|||||||
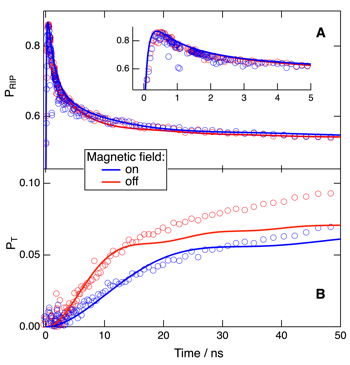
The dynamics of the ion pairs produced upon fluorescence quenching of the electron donor 9,10-dimethylanthracene (DMeA) by phthalonitrile have been investigated in acetonitrile and tetrahydrofuran using transient absorption spectroscopy. Charge recombination to both the neutral ground state and the triplet excited state of DMeA is observed in both solvents. The relative efficiency of the triplet recombination pathway decreases substantially in the presence of an external magnetic field. These results were analyzed theoretically within the differential encounter theory, with the spin conversion of the geminate ion pairs described as a coherent process driven by the hyperfine interaction. The early temporal evolution of ion pair and triplet state populations with and without magnetic field could be well reproduced in acetonitrile, but not in tetrahydrofuran where fluorescence quenching involves the formation of an exciplex. A description of the spin conversion in terms of rates, i.e., incoherent spin transitions, leads to an overestimation of the magnetic field effect. | ||||||||
Download this list in a RIS file or a BIB file or a PDF file
Contact:
Eric Vauthey
Physical Chemistry Department - Sciences II - University of Geneva
30, Quai Ernest Ansermet - CH-1211 Geneva 4 (Switzerland)
© All rights reserved by Eric Vauthey and the University of Geneva
Design and code by Guillaume Duvanel