

« Back to all publications
Download this list in a RIS file or a BIB file or a PDF file
|
 |
|||||||
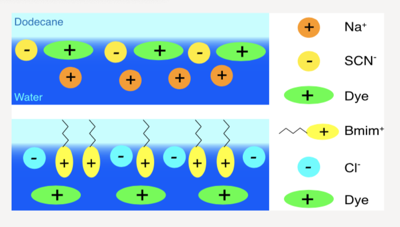
Interfaces with room-temperature ionic liquids (ILs) play key roles in many applications of these solvents, but our understanding of their properties is still limited. We investigate how the addition of ILs in the aqueous subphase affects the adsorption of the cationic dye malachite green at the dodecane/water interface using stationary and time-resolved surface second harmonic generation. We find that the interfacial concentration of malachite green depends crucially on the nature of both anionic and cationic constituents. This concentration reports on the overall charge of the interface, which itself depends on the relative interfacial affinity of the ions. Our results reveal that the addition of ILs to the aqueous subphase has similar effects to the addition of conventional salts. However, the IL cations have a significantly higher propensity to adsorb than small inorganic cations. Furthermore, the IL constituents show a synergistic effect, as the interfacial concentration of each of them also depends on the interfacial affinity of the other. | ||||||||



|
 |
|
|||||||
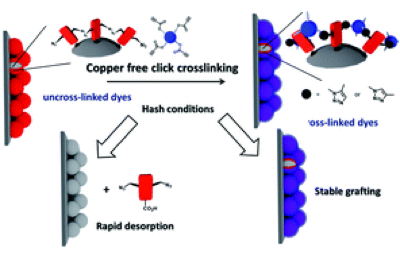
Naphthalenediimides (NDIs) are privileged scaffolds par excellence, of use in functional systems from catalysts to ion channels, photosystems, sensors, ordered matter in all forms, tubes, knots, stacks, sheets, vesicles, and colored over the full visible range. Despite this extensively explored chemical space, there is still room to discover core-substituted NDIs with fundamentally new properties: NDIs with cyclic trisulfides (i.e., trisulfanes) in their core absåorb at 668?nm, emit at 801?nm, and contract into disulfides (i.e., dithietes) upon irradiation at <475?nm. Intramolecular 1,5-chalcogen bonds account for record redshifts with trisulfides, ring-tension mediated chalcogen-bond-mediated cleavage for blueshifts to 492?nm upon ring contraction. Cyclic oligochalcogenides (COCs) in the NDI core open faster than strained dithiolanes as in asparagusic acid and are much better retained on thiol exchange affinity columns. This makes COC-NDIs attractive not only within the existing multifunctionality, particularly artificial photosystems, but also for thiol-mediated cellular uptake. | ||||||||
|
 |
|||||||
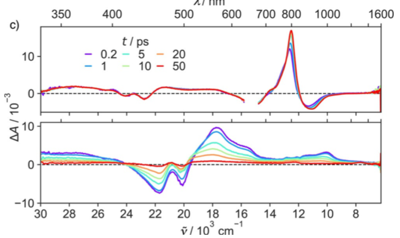
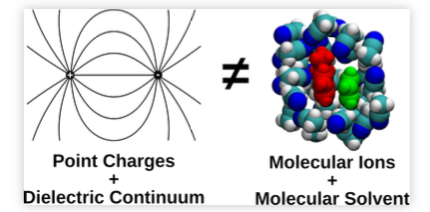
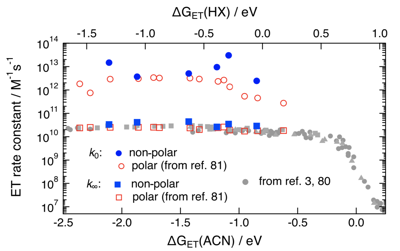
Reliable estimation of the driving force for photoinduced electron transfer between neutral reactants is of utmost importance for most practical applications of these reactions. The driving force is usually calculated from the Weller equation, which contains a Coulomb term, C, whose magnitude in polar solvents is debated. We have performed umbrella sampling molecular dynamics simulations to determine C from the potentials of mean force between neutral and ionic donor/acceptor pairs of different sizes in solvents of varying polarity. According to the simulations, C in polar solvents is a factor of 2 more negative than typically calculated according to the Weller equation. Use of the À-stack contact distance in the Weller equation instead of the van der Waals radius recovers the correct value of C, but this is mostly fortuitous due to the compensating effects of overestimating the dielectric screening at contact and neglecting both charge dilution and desolvation. | ||||||||
 |
|
|
|
|
 |
 |
|
|
||||||||
Design, synthesis and evaluation of push-pull N,N′-diphenyl-dihydrodibenzo[a,c]phenazines are reported. Consistent with theoretical predictions, donors and acceptors attached to the bent mechanophore are shown to shift absorption maxima to either red or blue, depending on their positioning in the chromophore. Redshifted excitation of push-pull fluorophores is reflected in redshifted emission of both bent and planar excited states. The intensity ratios of the dual emission in more and less polar solvents imply that excited-state (ES) planarization decelerates with increasing fluorophore macrodipole, presumably due to attraction between the wings of closed papillons. ES planarization of highly polarisable papillons is not observed in lipid bilayer membranes. All push-pull papillon amphiphiles excel with aggregation-induced emission (AIE) from bent ES as micelles in water and mechanosensitivity in viscous solvents. They are not solvatochromic and only weakly fluorescent (QY < 4%). | ||||||||
|
 |
 |
|
|||||||
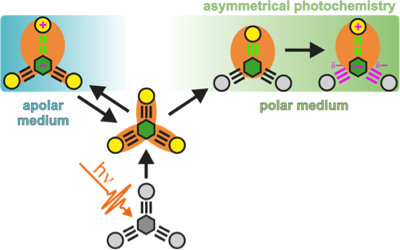
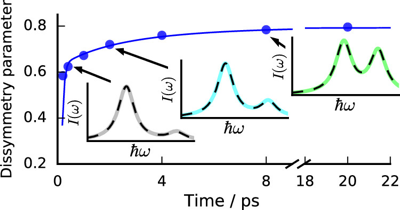
The nature of the electronic excited state of many symmetric multibranched donor–acceptor molecules varies from delocalized/multipolar to localized/dipolar depending on the environment. Solvent-driven localization breaks the symmetry and traps the exciton in one branch. Using a combination of ultrafast spectroscopies, we investigate how such excited-state symmetry breaking affects the photochemical reactivity of quadrupolar and octupolar A(-Ï€-D)2,3 molecules with photoisomerizable A-Ï€-D branches. Excited-state symmetry breaking is identified by monitoring several spectroscopic signatures of the multipolar delocalized exciton, including the S2â†S1 electronic transition, whose energy reflects interbranch coupling. It occurs in all but nonpolar solvents. In polar media, it is rapidly followed by an alkyne-allene isomerization of the excited branch. In nonpolar solvents, slow and reversible isomerization corresponding to chemically-driven symmetry breaking, is observed. These findings reveal that the photoreactivity of large conjugated molecules can be tuned by controlling the localization of the excitation. | ||||||||
|
 |
|||||||
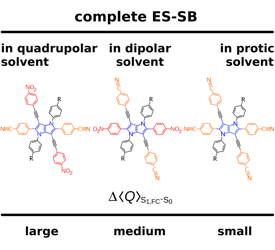
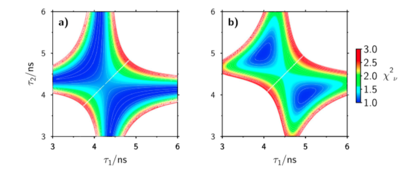
A significant number of quadrupolar dyes with a D-Ï€-A-Ï€-D or A-Ï€-D-Ï€-A structure, where D and A are electron donor and acceptor groups, were shown to undergo symmetry breaking (SB) upon optical excitation. During this process, electronic excitation, originally distributed evenly over the molecule, concentrates on one D−À–A branch, and the molecule becomes dipolar. This process can be monitored by time-resolved infrared spectroscopy and causes significant spectral dynamics. A theoretical model of excited-state SB developed earlier (Ivanov, A. I. J. Phys. Chem. C2018,122, 29165–29172) is extended to account for the temporal changes taking place in the IR spectrum upon SB. This model can reproduce the IR spectral dynamics observed in the -C≡C- stretching region with a D-Ï€-A-Ï€-D dye in two polar solvents using a single set of molecular parameters. This approach allows estimating the degree of asymmetry of the excited state in different solvents and its change during SB. Additionally, the relative contribution of different mechanisms responsible for the splitting of the symmetric and antisymmetric -C≡C- stretching bands, which are both IR active upon SB, can be determined. | ||||||||
Download this list in a RIS file or a BIB file or a PDF file
Contact:
Eric Vauthey
Physical Chemistry Department - Sciences II - University of Geneva
30, Quai Ernest Ansermet - CH-1211 Geneva 4 (Switzerland)
© All rights reserved by Eric Vauthey and the University of Geneva
Design and code by Guillaume Duvanel