

« Back to all publications
Download this list in a RIS file or a BIB file or a PDF file
|
 |



 |
|
|
 |
 |
|
|||||||
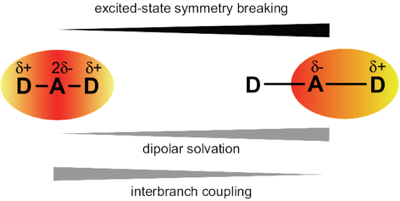
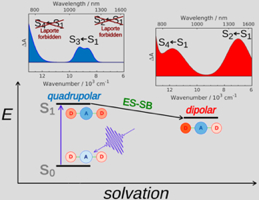
Excited-state symmetry breaking is investigated in a series of symmetric 9,10-dicyanoanthracenes linked to electron-donating groups on the 2 and 6 positions via different spacers, allowing for a tuning of the length of the donor-acceptor branches. The excited-state properties of these compounds are compared with their dipolar single-branch analogues. The changes in electronic structure upon their optical excitation are monitored by transient electronic spectroscopy in the visible and near-infrared regions as well as by transient vibrational spectroscopy in the mid-infrared. Our results reveal that, with the shortest branches, electronic excitation remains distributed almost symmetrically over the molecule even in polar environments. Upon increasing the donor–acceptor distance, excitation becomes unevenly distributed and, with the longest one, it fully localises on one branch in polar solvents. The influence of the branch length on the propensity of quadrupolar dyes to undergo excited-state symmetry breaking is rationalised in terms of the balance between interbranch coupling and solvation energy. | ||||||||
|
|
 |
|||||||
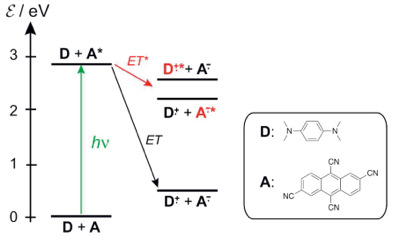
Excited-state symmetry breaking (ES-SB) is common to a large number of multibranched electron donor-acceptor (DA) molecules in polar environments. During this process, the electronic excitation, originally evenly distributed over the molecule, localizes, at least partially, on one branch. Due to the absence of an unambiguous spectroscopic signature in the UV-vis region, electronic transient absorption (TA) has not been the method of choice for real-time observation of this phenomenon. Herein, we demonstrate that the Laporte rule, which states that one-photon transitions conserving parity are forbidden in centrosymmetric molecules, provides such clear signature of ES-SB in electronic TA spectroscopy. Using a dicyanoanthracene-based D-A-D dye, we show that transitions from the S1 state of this molecule, which are initially Laporte forbidden, become allowed upon ES-SB. This leads to the rise of new TA bands, whose intensity provides a direct measure of the extent of asymmetry in the excited state. | ||||||||
|
||||||||
A simultaneous combination of porosity and tunable optoelectronic properties, common in covalent organic frameworks, is rare in shape-persistent organic cages. Yet, organic cages offer important molecular advantages such as solubility and modularity. Herein, we report the synthesis of a series of chiral imine organic cages with three built-in rylene units by means of dynamic imine chemistry and we investigate their textural and optoelectronic properties. Thereby we demonstrate that the synthesized rylene cages can be reversibly reduced at accessible potentials, absorb from UV up to green light, are porous, and preferentially adsorb CO2 over N2 and CH4 with a good selectivity. In addition, we discovered that the cage incorporating three perylene-3,4:9,10-bis(dicarboximide) units displays an efficient delayed fluorescence. Time-correlated single photon counting and transient absorption spectroscopy measurements suggest that the delayed fluorescence is likely a consequence of a reversible intracage charge-separation event. Rylene cages thus offer a promising platform that allows combining the porosity of processable materials and photochemical phenomena useful in diverse applications such as photocatalysis or energy storage. | ||||||||
 |
|
|||||||
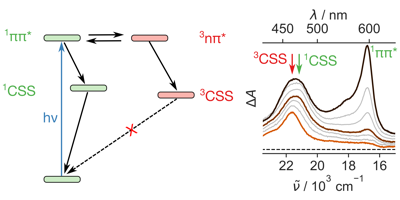
1,4,5,8-Naphthalenediimides (NDIs) are widely used motifs to design multichromophoric architectures due to their ease of functionalisation, their high oxidative power and the stability of their radical anion. The NDI building block can be incorporated in supramolecular systems by either core or imide functionalization. We report on the charge-transfer dynamics of a series of electron donor–acceptor dyads consisting of a NDI chromophore with one or two donors linked at the axial, imide position. Photo-population of the core-centred À–À* state is followed by ultrafast electron transfer from the electron donor to the NDI. Due to a solvent dependent singlet–triplet equilibrium inherent to the NDI core, both singlet and triplet charge-separated states are populated. We demonstrate that long-lived charge separation in the triplet state can be achieved by controlling the mutual orientation of the donor–acceptor sub-units. By extending this study to a supramolecular NDI-based cage, we also show that the triplet charge-separation yield can be increased by tuning the environment. | ||||||||
|
 |
|||||||
This study addresses a practical aspect of hybrid dye-sensitized photoelectrochemical cells by exploring a simple method to prepare multicomponent systems. Building on a previously reported methodology based on a copper-free click chemistry dipolar cycloaddition of azide with activated alkyne, a naphthalene diimide (NDI) derivative substituted with two propiolic esters was clicked on a NiO photocathode already coated with a diketopyrrolopyrrole (DPP) dye bearing two azido groups. A detailed photophysical study by transient absorption spectroscopy demonstrates that optical excitation of DPP dye leads to an effective electron transfer chain from the NiO valence band to the NDI passing via the DPP dye, resulting in a long-lived charge-separated state (hole in NiO/NDI radical anion) of 170 μs. The p-type dye-sensitized solar cells were also fabricated with the above molecular components and confirm the occurrence of the electron transfer as the performances of the solar cells were improved in terms of Voc and Jsc compared to the DPP dye lacking the NDI unit. The above-clicked system was also compared to a covalently linked DPP–NDI dyad, whose performances are 30% superior to the clicked system probably due to longer mean distance between the NiO surface and the NDI with the dyad. This finding paves the way for the design of multicomponent hybrid dye-sensitized photoelectrochemical cells by chemistry on the electrode. | ||||||||
 |
|
|||||||
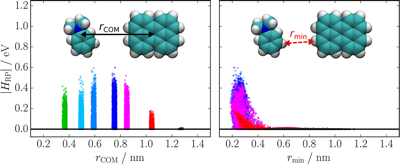
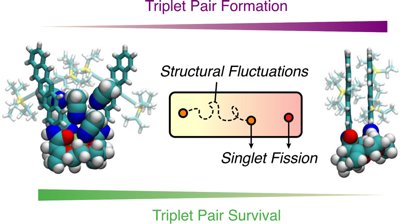
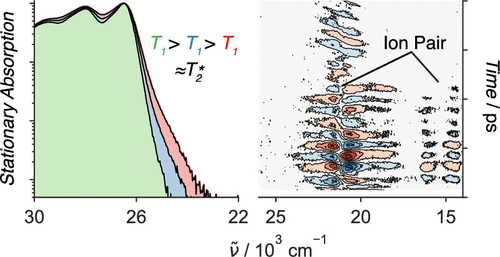
Singlet fission (SF), i.e., the splitting of a high-energy exciton into two lower-energy triplet excitons, has the potential to increase the efficiency for harvesting spectrally broad light. The path from the photopopulated singlet state to free triplets is complicated by competing processes that decrease the overall SF efficiency. A detailed understanding of the whole cascade and the nature of the photoexcited singlet state is still a major challenge. Here, we introduce a pentacene dimer with a flexible crown ether spacer enabling a control of the interchromophore coupling upon solvent-induced self-aggregation as well as cation binding. The systematic change of solvent polarity and viscosity and excitation wavelength, as well as the available conformational phase space, allows us to draw a coherent picture of the whole SF cascade from the femtosecond to microsecond time scales. High coupling leads to ultrafast SF (<2 ps), independent of the solvent polarity, and to highly coupled correlated triplet pairs. The absence of a polarity effect indicates that the solvent coordinate does not play a significant role and that SF is driven by intramolecular modes. Low coupling results in much slower SF (∼500 ps), which depends on viscosity, and leads to weakly coupled correlated triplet pairs. These two triplet pairs could be spectrally distinguished and their contribution to the overall SF efficiency, i.e., to the population of free triplets, could be determined. Our results reveal how the overall SF efficiency can be increased by conformational restrictions and control of the structural fluctuation dynamics. | ||||||||
|
||||||||
Although biological imaging is mostly performed in aqueous media, it is hardly ever considered that water acts as a classic fluorescence quencher for organic fluorophores. By investigating the fluorescence properties of 42 common organic fluorophores recommended for biological labelling, we demonstrate that H2O reduces their fluorescence quantum yield and lifetime by up to threefold and uncover the underlying fluorescence quenching mechanism. We show that the quenching efficiency is significantly larger for red-emitting probes and follows an energy gap law. The fluorescence quenching finds its origin in high-energy vibrations of the solvent (OH groups), as methanol and other linear alcohols are also found to quench the emission, whereas it is restored in deuterated solvents. Our observations are consistent with a mechanism by which the electronic excitation of the fluorophore is resonantly transferred to overtones and combination transitions of high-frequency vibrational stretching modes of the solvent through space and not through hydrogen bonds. Insight into this solvent-assisted quenching mechanism opens the door to the rational design of brighter fluorescent probes by offering a justification for protecting organic fluorophores from the solvent via encapsulation. | ||||||||
|
 |
|||||||
The absorption band shape of chromophores in liquid solution at room temperature is usually dominated by pure electronic dephasing dynamics, which occurs on the sub-100 fs time scale. Herein, we report on a series of dyads consisting of a naphthalenediimide (NDI) electron acceptor with one or two phenyl-based donors for which photoinduced intramolecular electron transfer is fast enough to be competitive with pure electronic dephasing. As a consequence, the absorption band of the π-π* transition of these dyads is broader than that of the NDI alone to an extent that scales with the electron transfer rate. Additionally, this reaction is so fast that it leads to the impulsive excitation of a low-frequency vibrational mode of the charge-separated product. Quantum-chemical calculations suggest that this vibration involves the C-N donor-acceptor bond, which shortens considerably upon electron transfer. | ||||||||
Download this list in a RIS file or a BIB file or a PDF file
Contact:
Eric Vauthey
Physical Chemistry Department - Sciences II - University of Geneva
30, Quai Ernest Ansermet - CH-1211 Geneva 4 (Switzerland)
© All rights reserved by Eric Vauthey and the University of Geneva
Design and code by Guillaume Duvanel