

« Back to all publications
Download this list in a RIS file or a BIB file or a PDF file
|
 |
|||||||
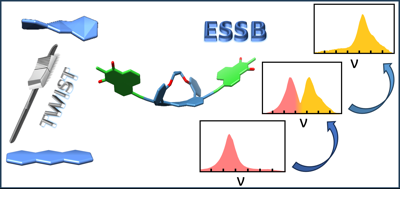
In electron donor-acceptor (D-A) molecules, the relative orientation of constituents has a dramatic influence over their performance. However, the D and A subunits are generally composed of planar aromatic backbones, and the effect of curvature is rarely explored. Here, we investigate how the twist of the aromatic core of a symmetric double-branched D-π-A molecule affects the nature and dynamics of its lower singlet excited state. We show that the twisting of the central donor not only affects the chiroptical properties, and increases the triplet yield, but also facilitates excited-state symmetry breaking (ESSB) and the trapping of the exciton on one D-π-A branch of the molecule. This enhancement is attributed to the decrease in the interbranch coupling upon distortion. Because of this, the loss of the coupling upon ESSB requires a smaller gain in solvation energy to be compensated for and, thus, exciton trapping occurs in a less polar solvent. Consequently, distortion can be viewed as an additional tuning knob for controlling the localisation of electronic excitation in large conjugated systems. | ||||||||



 |
|
|||||||
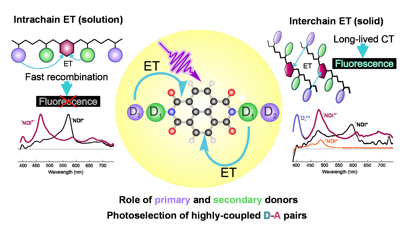
lectron donor–acceptor (D–A) polymers are emerging as promising candidates for the development of solid materials with tunable emission. Herein, we investigate the excited-state dynamics of polymers consisting of a central naphthalenediimide (NDI) acceptor with two polystyrene donor chains and copolymers with various secondary donors incorporated. We find strong differences in the dynamics when going from diluted polymer solutions to pure polymer films. In liquids, ultrafast intrachain electron transfer from a styrenic donor to the excited NDI, followed by sub-nanosecond charge recombination to the ground state is observed. Because of the tight packing in the film, ultrafast electron transfer occurs between donors and acceptors of different polymer chains. Emission is found to originate from the most strongly coupled D–A pairs, for which electron transfer is so fast that it leads to a lifetime broadening of the NDI absorption band. Because of this, these highly coupled pairs can be photoselected upon red-edge excitation. The charge-transfer state decays on the tens of ns timescale via radiative and non-radiative charge recombination to the ground state as well as via charge recombination to the triplet state of NDI. This latter pathway, which is detrimental to the fluorescence quantum yield, is almost suppressed with the strongest secondary donor. Finally, we show that excitation of the secondary donor instead of the NDI acceptor does not lead to the population of the charge-transfer state and thus does not contribute to the luminescence of the films. | ||||||||
Download this list in a RIS file or a BIB file or a PDF file
Last modified February 11 2026
Contact:
Eric Vauthey
Physical Chemistry Department - Sciences II - University of Geneva
30, Quai Ernest Ansermet - CH-1211 Geneva 4 (Switzerland)
© All rights reserved by Eric Vauthey and the University of Geneva
Design and code by Guillaume Duvanel