Recherche (en anglais)
The central theme of the research in our group is the application of computational methods to study the structure, reactivity, and electronic properties of organic and bio-organic systems.
The research in the field of bio-organic chemistry concentrates on the modeling of conformational behavior, properties as well as on investigations of the relationship between the structure and function of complex supramolecular systems which are studied by quantum chemistry methods and by molecular dynamics simulations. The focus is also on applications of computational methods to understand mechanism of organic reactions, and on the theoretical studies of reactive intermediates.
The key points of our research projects include:
- Different types of non-covalent interactions are studied in general by DFT and ab-initio methods with a specific focus on interactions such as π-π, anion-π, halogen bond, etc.
- Interactions between anions and polycyclic π-systems are explored in the context of ion transport, sensing or organocatalysis.
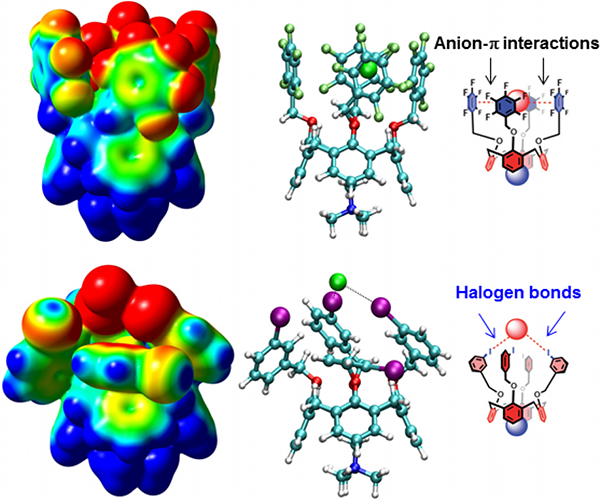
Example of calix[4]arene anion transporters, where the function is based on the anion-π interaction (top). After a minimal structural modification (meta-iodo substitution of arenes) the DFT modeling reveals two halogen bonds being responsible for the function (bottom). The transporting aptitude through the membrane was demonstrated on experimental grounds by Prof. Matile group.
- Modeling of properties, conformational behavior, and interactions in complexes between organic substrates and proteins such as streptavidin.
- Modeling of π-π interactions in acceptor-acceptor and donor-acceptor complexes using quantum chemistry methods.
- Molecular modeling of conformational behavior, properties and function of multifunctional supramolecular assemblies such as rigid-rod β-barrels, rigid π-rods, anion π-slides, etc.

The combination of DFT and MD simulations show the aptitude of strategically designed rigid-rod β-barrel to form high affinity complexes with an analyte (green CPK structure). This supramolecular structure was shown by Matile group to be a very selective and sensitive sensor, that could be assimilated to the artificial tongue, able to detect for example the L-glutamate (active ingredient of soy sauce) in micromolar concentrations.
- Molecular dynamics simulations as well as molecular mechanics calculations of complex supramolecular architectures based on π-π interactions.

Naphthalenediimide (NDI) π-π complexes were studied by DFT, ab-initio and MD simulations in context of zipper assemblies. In such supramolecular structures the NDIs are attached to the p-oligo-phenyl or oligo-phenylethynyl rigid rods, which assemble via interdigitation to form the zipper assemblies held together by a multitude of non-covalent interactions.
- Computational investigations of the mechanism of organic reactions and their selectivity for cycloadditions, cyclizations, and other useful organic transformations. Emphasis is also on reactions involving reactive intermediates.
- Stabilization of carbocations and hypervalent species by π-systems, investigated using high level ab-initio and DFT methods.
- Computational methods are used to enhance our understanding of stereoselectivity and improve the enantioselectivity of synthetically useful reactions.

Schematic energy profile for the preferred asymmetric conjugate addition to the Si-face of the E-enamine, together with the optimized TS structure (right). Useful Michael adducts are obtained in this transformation catalyzed by N-iPr-S-bipyrrolidine catalysts.
These computational investigations are often done in collaboration with groups of Profs. S. Matile, A. Alexakis, E. Vauthey, Ch. A. Schalley, P. Metrangolo, ..., where the systems under investigation are synthesized and their properties or function are evaluated by experimental methods.