Analysis of translatome by Ribo-Seq (Ribosome Profiling)
Ribosome profiling, or Ribo-Seq (also named ribosome footprinting), is a technique that uses specialized messenger RNA (mRNA) sequencing to determine which mRNAs are being actively translated (1,2). Ribo-Seq produces a “global snapshot” of all the ribosomes active in a cell at a particular moment, known as a translatome. Ribo-Seq targets only mRNA sequences protected by the ribosome during the process of decoding by translation unlike RNA-Seq, which sequences all of the mRNA of a given sequence present in a sample.
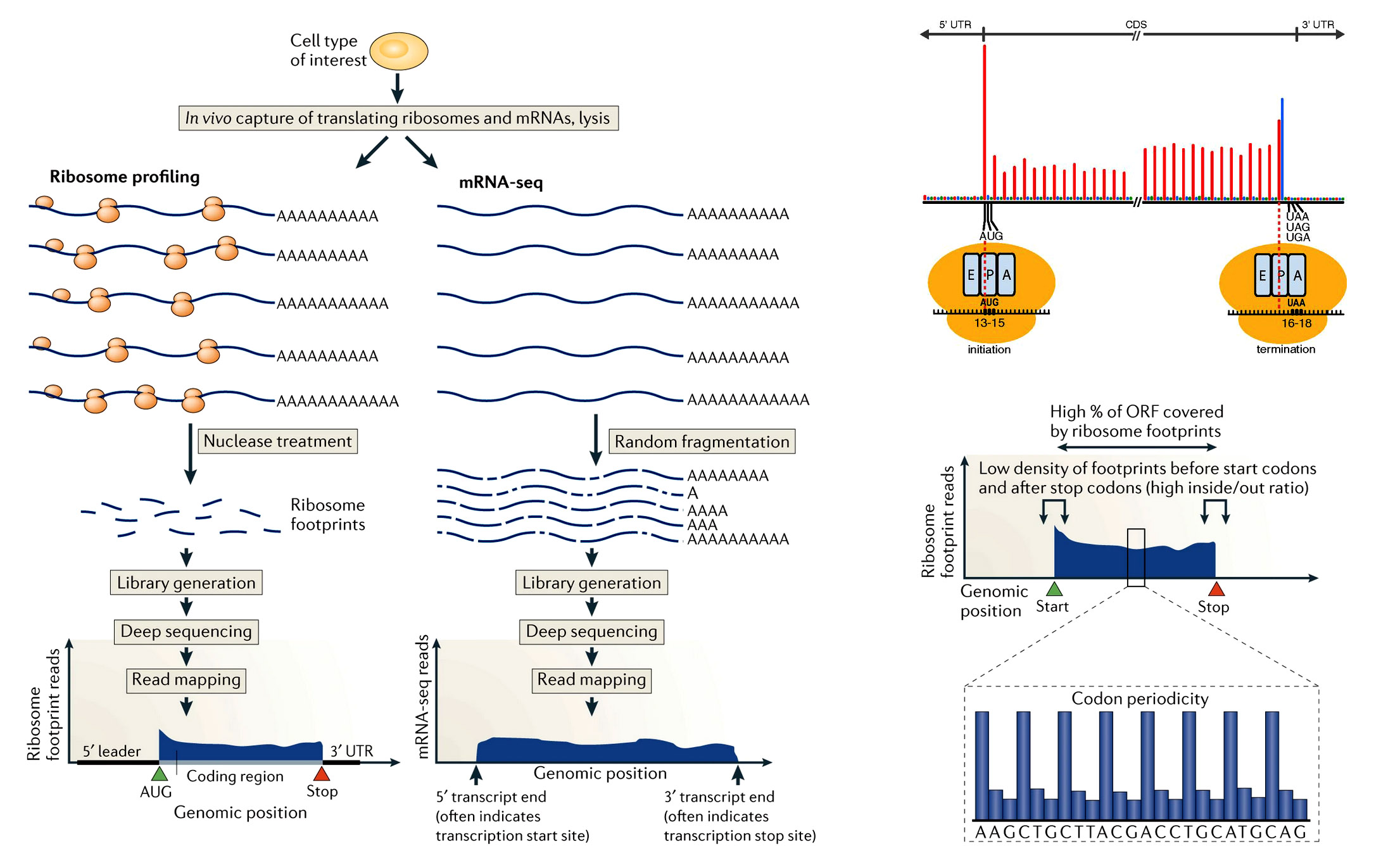
Fig. 1. An overview of Ribo-Seq (ribosome profiling) (2).
In Ribo-Seq ribosome-bound mRNAs are treated with nuclease, resulting in mRNA protected fragments (RPFs) termed ‘footprints’. These RPFs are isolated and converted to a library for deep sequencing. The RPF typically show precise positioning between the start and the stop codon of a gene, which facilitates global and experimental identification of genomic coding regions. By comparison, mRNA sequencing (mRNA-seq) captures random fragments covering the entire mRNA transcript. The positional information determined by standard mRNA-seq allows approximate determination of transcript boundaries, but it is less precise than that collected by Ribo-Seq, owing to the loss of 5ʹ and 3ʹ ends during the fragment generation method that is typically used.
There are three main uses of Ribo-Seq: identifying translated mRNA regions, observing how nascent peptides are folded, and measuring the amount of specific proteins that are synthesized.
- 1) Ribo-Seq enables researchers to identify the location of translation start sites, the complement of translated ORFs in a cell or tissue, the distribution of ribosomes on a messenger RNA, and the speed of translating ribosomes.
- 2) Coupling ribosome profiling with the other techniques such as IP or ChIP can elucidate how and when newly synthesized proteins are folded. Using the footprints provided by Ribo-Seq, specific ribosomes associated with factors, like chaperones, can be purified.
- 3) Ribo-Seq can also be used to estimate translation efficiency, a proxy for protein synthesis. For this application, Ribo-Seq and matched RNA-Seq data are generated. Translation efficiency can then be computed as the ribosome occupancy of each gene while controlling for its RNA expression. This approach can be coupled with mass spectrometry methods the analyzed proteome.

Fig. 2. Qualitative and quantitative data provided by Ribo-Seq (ribosome profiling) (2).
A diverse sample pool of mRNAs, distinguished by colour, is shown, together with a corresponding representative genome browser plot of Ribo-Seq and RNA-Seq data derived from this pool. Ribo-Seq facilitates experimental determination of translated regions, including short open reading frames (sORFs), and upstream ORFs (uORFs). Pausing during translation elongation may result in peaks in ribosome footprint reads within ORFs. Transcript abundances may not correlate closely with the instantaneous protein synthesis rates. The collection of quantitative data for both transcript abundances and protein synthesis rates enables the relative translation efficiencies to be inferred. These can vary over several orders of magnitude within a given organism in a given state. The translation efficiency can also change over time for a given mRNA, reflecting dynamic regulation at the level of translation.
1. N.T. Ingolia , S. Ghaemmaghami, J.R. Newman, J.S. Weissman (2009), Genome-wide analysis in vivo of translation with nucleotide resolution using ribosome profiling. Science 324, 218-223.
2. G.A. Brar, J.S. Weissman (2015), Ribosome profiling reveals the what, when, where and how of protein synthesis. Nat Rev Mol Cell Biol 16, 651-664.