[646] Smooth muscle cells in the pathophysiology of vascular disease
|
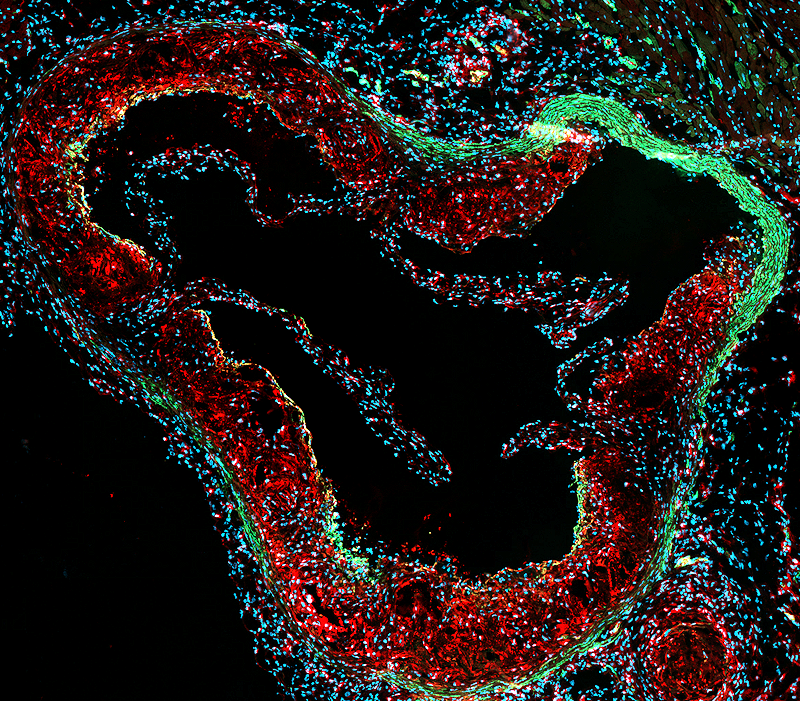
Atherosclerosis, the main cause of cardiovascular disease, is a pathological process that leads to arterial lumen narrowing and occlusion. During this process, smooth muscle cells (SMC) accumulate into the intima, where they switch from a contractile to a synthetic phenotype. They were classically considered as beneficial players by contributing to the fibrous cap formation and plaque stability. However, SMC can acquire pro-inflammatory macrophage-like phenotypes, contributing to atherosclerotic plaque instability. Our research line is focused on the identification of factors and mechanisms responsible for the detrimental SMC phenotypic changes. We identified S100A4 as a marker of the synthetic SMC phenotype. The extracellular form of S100A4 induces a SMC transition toward a pro-inflammatory phenotype. Noteworthy, extracellular S100A4 neutralization promotes atherosclerotic plaque stabilization in ApoE-/- mice. We are investigating the mechanisms involved in S100A4-induced SMC pro-inflammatory phenotypic transition as well as the possible activation of monocytes by SMCs. To decipher the contribution of systemic and SMC-specific S100A4 in SMC phenotypic plasticity during atherosclerosis, we are using a full-S100A4 KO mouse model and are setting up a SMC lineage tracing mouse model associated with SMC-specific deletion of S100A4. The ultimate aim of our work is the development of tools to influence the evolution of atherosclerotic lesions and hence reduce adverse outcome of atherosclerosis on clinical events. Our studies should be instrumental in unveiling mechanisms responsible for the detrimental pro-inflammatory SMC phenotypic changes in atherosclerosis as well as the possible activation of monocytes by SMCs. This is a growing research field in cardiovascular disease that challenges our classical understanding of the pathogenesis of atherosclerosis, with new therapeutic perspectives. |
publications du groupe