A new pipeline for metagenomic analyses
The number of microorganisms living in our digestive tracts is enormous. Traditionally, bacterial cultivations were used for isolation and classification of many bacterial strains. These approaches however are limited only to culturable bacteria allowing low-resolution microbial community fingerprinting. Metagenomic sequencing holds great promise for high-resolution strain and subspecies analyses, bypassing the need for lab cultivation of individual organisms in a petri dish. Still, vast majority of bacterial genomes remain unclassified, and there is no information of the microbiota on a sub-species level, largely owing to the lack of bioinformatics tools that would enable analysis of the huge amount of sequencing data.
The ATLAS pipeline
In a recent paper in BMC Bioinformatics, Silas Kieser from Pr. Mirko Trajkovski laboratory, in collaboration with Pr. Evgeny Zdobnov laboratory and with researchers from the Pacific Northwest National Laboratory developed a new user-friendly tool called metagenome-atlas allowing analysis of large amounts of data. Metagenome-atlas fills an important analysis gap by combining the key state-of-the-art tools together. This open-source software is easy to install and use: scientists can run metagenomic analyses with only 3 command lines.
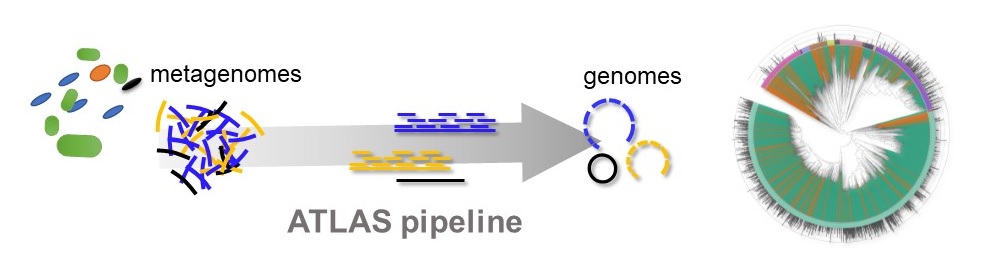
Schematic representation of the ATLAS pipeline that allows processing raw genetic sequences (metagenomes) and translating them into number of microorganisms (genomes). © unige – Silas Kieser
A tried and tested tool
This new tool is already used by researchers from Pr. Mirko Trajkovski laboratory for investigating the mouse gut microbiome and its similarities to the human counterpart. It allowed them to identify new bacterial families, as well as hundreds of species and subspecies not detectable with traditional lab cultivation. Metagenome-atlas is already in use by scientists around the world for studying different microbial communities ranging from gut to freshwater ecosystems.
6 Jul 2020